


AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model
들어가며
2025년 6월 25일, 구글 딥마인드에서 기능유전체학(functional genomics) 분야의 새로운 foundation model, 알파게놈(AlphaGenome)을 발표했습니다. AlphaGenome은 오직 DNA 서열만을 이용하여 유전자 발현량, 스플라이싱(splicing) 패턴, 염색질 접근성(chromatin accessibility) 등 다양한 functional genomic modality를 예측하는 “sequence-to-function” 모델로서, 유전 변이에 의해 유전체 상에 일어나는 다양한 기능적인 변화들을 한번에, 그리고 높은 정확도로 예측할 수 있다는 점에서 유전체 분석 분야에서 폭넓은 활용도를 보일 것으로 기대되는 모델입니다. 이 포스팅에서는 AlphaGenome preprint를 살펴보고, 모델의 특징과 실험 결과를 정리해보면서 이 모델이 생물정보학 분야에서 과연 어떤 의미를 가지는지 알아보도록 하겠습니다.
이에 앞서, AlphaGenome의 작동 방식과 의의를 이해하기 위한 필수 배경 지식에 대해 먼저 짚고 넘어가볼까요?
유전체
우리 인간의 몸은 세포라는 단위로 구성되어 있고, (거의 모든) 세포의 안에는 막으로 둘러싸인 핵이라는 세포 소기관이 존재합니다. 핵 내에는 A, C, G, T의 4개 염기가 이어붙여진 DNA라는 길다란 분자들이 존재합니다. DNA는 핵 내에 흐물흐물하게 퍼져서 존재하는 것이 아니라, “히스톤”이라는 공 모양의 단백질에 감긴 상태로 존재하는데요, 마치 수많은 실타래에 실이 감겨있는 형태로 볼 수 있습니다. 이렇게 DNA와 히스톤 단백질 및 여타 단백질들의 복합 구조를 염색질(chromatin)이라고 하며, 염색질은 세포의 상태에 따라, 혹은 DNA 가닥의 위치에 따라 느슨하게 풀리기도 하고 단단하게 결합하여 압축되기도 합니다. 가장 극단적으로 압축된다면, 우리가 언젠가 한 번쯤 현미경으로 보았을 염색체(아래 그림)의 모양을 띠게 됩니다.

DNA, 염색질, 염색체. 출처: https://www.genome.gov/genetics-glossary/Chromatin
DNA가 세포 내에 어떻게 구조화되어 저장되어 있는지 간단히 알아보았습니다. 그렇다면 유전체(genome)란 대체 무엇일까요? 이를 위해서는 DNA가 담고 있는 정보에 대한 이해가 필요합니다.
유전자(gene)
DNA는 단순히 A, C, G, T라는 네 개의 염기가 무작정 나열된 것이 아닙니다. A, C, G, T의 나열, 즉 염기 서열에는 생명체가 살아가는 데 필요한 모든 정보가 담겨 있습니다. 마치 책 안의 글자들이 하나하나 모여 고유의 뜻을 가지는 단어들을 이루고, 단어들이 모여 문장을 구성하며, 문장들이 모여 하나의 완전한 이야기를 만들어내는 것처럼요.
DNA 서열 중에서 특정한 기능을 하는 단백질을 만들어내는 정보를 담고 있는(정보를 ‘암호화’한다고 합니다) 구간을 우리는 유전자(gene)라고 부릅니다. 하나의 유전자는 보통 수백 개에서 수만 개의 염기로 이루어져 있으며, 각 유전자는 고유한 단백질을 만들어내는 설계도, 혹은 레시피와 같은 역할을 합니다. 예를 들어, 우리 눈의 색깔을 결정하는 멜라닌 색소를 만드는 단백질의 정보가 담긴 유전자가 있고, 혈액 속에서 산소를 운반하는 헤모글로빈 단백질의 정보가 담긴 유전자도 따로 존재하는 식이죠.
유전체(genome)
유전체란 한 생물이 가지고 있는 모든 유전 정보를 모두 합친 것을 총체적으로 의미합니다. 특히, 단백질에 대한 정보를 직접적으로 담고 있는 암호화 부위(coding region)는 물론이거니와, 아직 그 기능이 명확히 밝혀지지 않은 비암호화 부위(non-coding region)들을 모두 포함하는 개념이라는 것이 중요합니다. 인간의 경우, 아버지와 어머니로부터 각각 23개씩 물려받은 총 46개 염색체에 담긴 약 32억 개의 염기쌍이 바로 인간의 유전체를 구성하게 됩니다.
유전체 안에는 앞서 설명한 유전자들이 약 2만 개 정도 존재한다고 알려져 있습니다. 하지만 흥미롭게도, 인간의 DNA 전체에서 실제로 단백질을 만드는 정보를 담고 있는 암호화 부위의 비중은 전체의 약 2% 정도에 불과합니다. 나머지 98%의 비암호화 부위들이 과연 어떤 역할을 하는지에 대해서는 아직도 활발한 연구가 진행되고 있고, 흔히 유전자의 발현을 조절하거나 염색질의 구조를 유지하는 등 다양한 기능을 수행하는 것으로 알려져 있습니다.
AlphaGenome은 바로 이러한 비암호화 부위의 기능과, 그 안에 숨겨진 규칙들을 이해하고자 하는 모델이며, 나아가 비암호화 부위의 DNA 염기 하나하나가 “변했을 때” 어떤 일들이 생길지를 예측하는 모델입니다.
유전 변이(variant; mutation)
그렇다면 DNA 염기가 "변한다"는 것은 무엇을 의미할까요? 이를 이해하기 위해서는 유전 변이(variant 혹은 mutation)라는 개념을 이해해야 합니다.
유전 변이란 기준이 되는 DNA 서열(보통 참조유전체(reference genome)라 부르는)과 비교했을 때, 특정 위치의 염기가 다르게 나타나는 현상을 말합니다. 예를 들어, 어떤 위치에서 참조 서열에서는 A 염기가 있는데, 실제로는 T 염기가 관찰된다면 이것이 바로 유전 변이인 것이죠. 마치 책을 베껴 쓰는 과정에서 가끔 오타가 생기는 것과 비슷하다고 볼 수 있습니다.
유전 변이는 크게 두 가지로 나눌 수 있습니다. 첫 번째는 암호화 부위에서 발생하는 coding variant입니다. 이러한 변이는 직접적으로 단백질의 아미노산 서열을 바꾸기 때문에 그 영향을 비교적 예측하기 쉽습니다. 단백질의 기능이 완전히 망가지거나, 반대로 전혀 영향을 주지 않는 경우가 대부분입니다.
두 번째는 비암호화 부위에서 발생하는 non-coding variant입니다. 이 경우가 훨씬 복잡한데, 직접적으로 단백질을 바꾸지는 않지만 유전자의 발현 정도를 조절하는 역할을 할 수 있습니다. 이를 양적 형질 유전자좌(QTL; Quantitative Trait Locus)라고 부르기도 하는데, 특정 형질(예: 키, 혈압, 질병 감수성 등)에 미치는 영향이 연속적이고 정량적으로 나타나는 특징이 있습니다. 바로 이 부분이 AlphaGenome이 주목하는 영역 중 하나입니다.
기능유전체
앞서 살펴본 유전체가 “생명의 설계도”라는 개념이었다면 기능유전체는 “그 설계도가 어떻게 사용되고 있는지”를 나타내는, 보다 동적인 개념입니다. 이번 섹션에서는 기능유전체 전반의 개념들에 대해 알아보도록 합시다.
프로모터(promoter)와 유전자 발현(gene expression)

프로모터와 유전자 발현. 출처: National Human Genome Research Institute
유전자가 단백질을 만들어내는 설계도라면, 그 설계도를 언제, 어느 정도로 사용할지를 결정하는 것은 바로 유전자 발현(gene expression)의 조절을 통해 이루어집니다. 유전자가 발현된다는 것은 결국, 짧은 시간동안 해당 유전자의 설계도 복사본이 마구 만들어져 실제로 단백질을 합성하는 세포 소기관에게 전달되어 단백질 합성이 이루어진다는 의미입니다. 이 유전자 발현 조절에 핵심적인 역할을 하는 것이 프로모터(promoter)라는 DNA 서열입니다.
프로모터는 각 유전자의 시작 부분 앞쪽에 위치한 조절 서열로, 기능적으로는 마치 "지금 이 유전자를 사용할지 말지" 결정하는 신호등과 같은 역할을 합니다. 프로모터 서열에는 RNA 중합효소(RNA polymerase)라는 효소가 결합하게 되고, 이 효소가 DNA를 읽어서 전령 RNA(messenger RNA, mRNA)로 전사(transcription)하는 과정이 시작됩니다. 프로모터의 활성도가 높을수록 더 많은 mRNA가 만들어지고, 결과적으로 더 많은 단백질이 생산되는 것이죠.
흥미롭게도, 같은 유전자라도 세포의 종류나 상황에 따라 발현 정도가 달라집니다. 예를 들어, 인슐린 유전자는 췌장의 베타 세포에서는 활발히 발현되지만, 뇌세포에서는 거의 발현되지 않습니다. 이러한 조절이 가능한 이유는 특정한 프로모터 서열을 인식하여 결합하는 다양한 종류의 조절 요소들이 존재하기 때문입니다.
전사 인자(transcription factor)
이러한 조절 요소 단백질들을 흔히 전사 인자(transcription factor, TF)라고 부릅니다.

출처: Wikipedia
전사 인자는 주로 프로모터에 존재하는 특정한 DNA 서열에 결합하여 유전자 발현을 촉진하거나 억제하는 단백질로, 인간에게는 약 1,600개 정도의 전사 인자가 존재한다고 알려져 있습니다. 특정 조합의 전사 인자의 작용에 의해 세포 타입별로, 그리고 발달 단계별로 고유한 유전자 발현 패턴을 만들어내고, 이러한 복잡한 조절 네트워크 덕분에 같은 유전체(동일한 유전 정보)를 가진 세포들이 뇌세포, 간세포, 근육세포 등 전혀 다른 기능을 수행할 수 있게 됩니다.
염색질 접근성(chromatin accessibility)
전사 인자가 DNA에 결합하려면 먼저 그 DNA 부위에 물리적으로 접근할 수 있어야 합니다. 하지만 앞서 설명했듯이 DNA는 히스톤 단백질에 감겨있는 염색질 형태로 존재하기 때문에, 모든 DNA 서열이 항상 접근 가능한 것은 아닙니다.
염색질 접근성(chromatin accessibility)이란 특정 DNA 서열이 전사 인자나 다른 단백질들이 결합할 수 있을 정도로 열려있는 상태를 의미합니다. 염색질이 느슨하게 풀려 열린 상태(open chromatin)에서는 전사 인자들이 쉽게 결합할 수 있어 유전자 발현이 활발해지고, 반대로 염색질이 단단히 압축되어 닫힌 상태(closed chromatin)에서는 유전자 발현이 억제됩니다.
염색질 접근성은 다양한 요인에 의해 조절됩니다. 크로마틴 리모델링 복합체(chromatin remodeling complex)라는 단백질 복합체들이 ATP 에너지를 사용해서 히스톤 단백질의 위치를 이동시키거나 제거하여 DNA를 노출시키기도 하고, 특정 전사 인자들이 "개척자 전사 인자(pioneer transcription factor)" 역할을 하여 닫힌 염색질을 여는 역할을 하기도 합니다.
인핸서(enhancer), 염색질 접촉(chromatin contact), 3차원 genome(3D genome)
한편, 유전자 발현 조절에서 프로모터만큼 중요한 또 다른 요소가 바로 인핸서(enhancer)입니다. 인핸서는 유전자로부터 수천에서 수백만 염기 떨어진 곳에 위치하면서도 해당 유전자의 발현을 촉진하는 DNA 서열입니다.

Carullo and Day (2019)
수천~수백만 염기씩이나 멀리 떨어진 인핸서가 어떻게 특정 유전자에 영향을 줄 수 있을까요? 이는 3차원 유전체 구조와 관련이 있습니다. DNA는 핵 내에서 단순히 일직선으로 펼쳐져 있는 것이 아니라, 복잡하게 접혀 고리(루프)를 형성하면서 3차원적인 구조를 만들어냅니다. 이 과정에서 선형적으로는 멀리 떨어져 있던 인핸서와 프로모터가 물리적으로 가까워져 직접 상호작용할 수 있게 되는 것이죠.
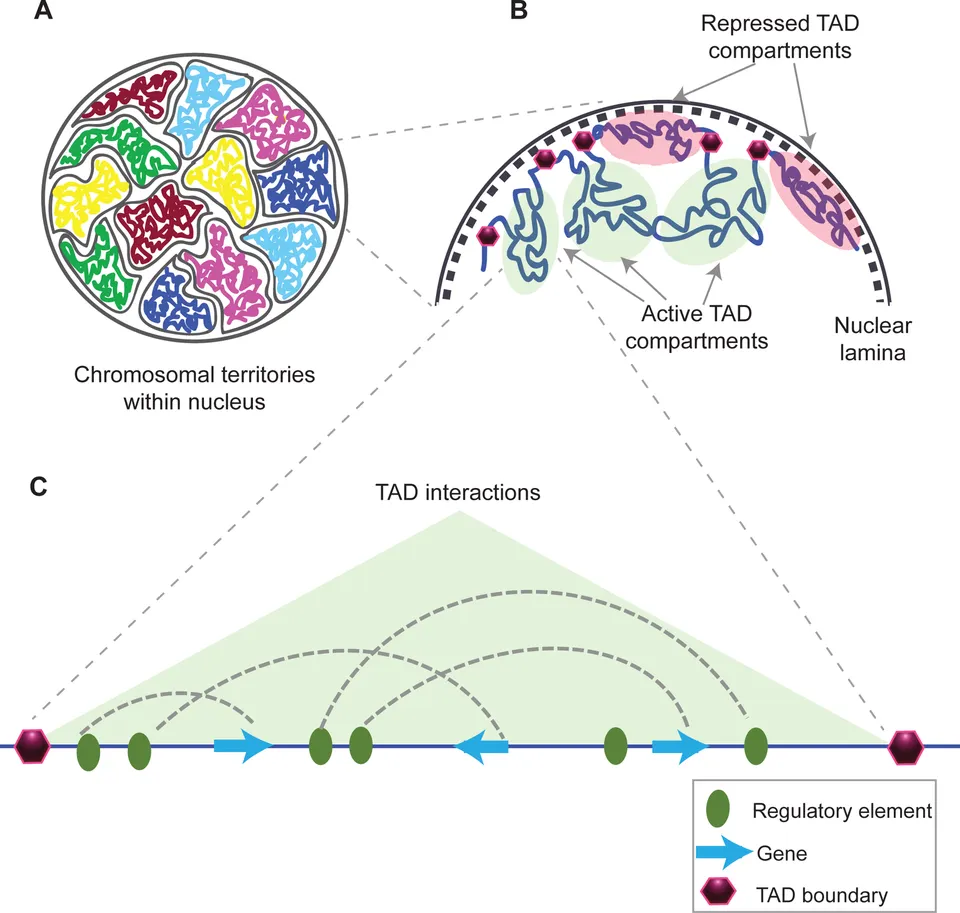
Topologically associating domain (TAD). 출처: Wikipedia
이러한 염색질 접촉(chromatin contact)은 무작위로 일어나는 것이 아닙니다. CTCF라는 단백질과 코헤신(cohesin) 복합체가 협력하여 특정 DNA 구간들을 루프 형태로 만들어주는 역할을 합니다. 이때 형성되는 구조를 위상적 연관 도메인(TAD; Topologically Associating Domain)이라고 하는데, 같은 TAD 내의 인핸서와 프로모터는 서로 상호작용할 확률이 높아집니다.
재밌는 것은, CTCF 단백질 또한 선호하는 DNA 서열이 있다는 점입니다. (”CCCTC-Binding Factor”라는 단백질 이름에서부터 알 수 있죠.) 3차원 유전체 구조 형성에 깊게 관여하는 CTCF 단백질이 선호하는 유전체 서열이 미리 정해져 있다는 사실은 “3차원 유전체 구조 형성 패턴” 그 자체도 유전체 내에 하나의 정보로서 암호화되어 있다는 것을 의미합니다. 놀랍지 않나요? (마치 ‘접는 선’이 미리 표시된 종이접기 종이 같다고나 할까요..!)
스플라이싱(splicing)
DNA에서 mRNA로 전사가 완료되었다고 해서 바로 단백질이 만들어지는 것은 아닙니다. 진핵생물의 유전자 서열은 대부분 엑손(exon)과 인트론(intron)이라는 서열이 반복적으로 배열되어 있는데, 여기서 엑손은 실제로 단백질 서열 정보를 담고 있는 부분이고, 인트론은 그 사이사이에 끼어있는 서열로서 최종 mRNA에서는 제거되어야 하는 부분입니다.

Splicing 과정의 모식도. 출처: Khan Academy.
스플라이싱(splicing)이란 인트론을 제거하고 엑손들만을 이어붙이는 과정을 말합니다. 이 과정은 스플라이소솜(spliceosome)이라는 거대한 RNA-단백질 복합체에 의해 수행되며, 매우 정교한 서열 인식 과정을 거쳐 정확한 위치에서 절단과 연결이 일어납니다.
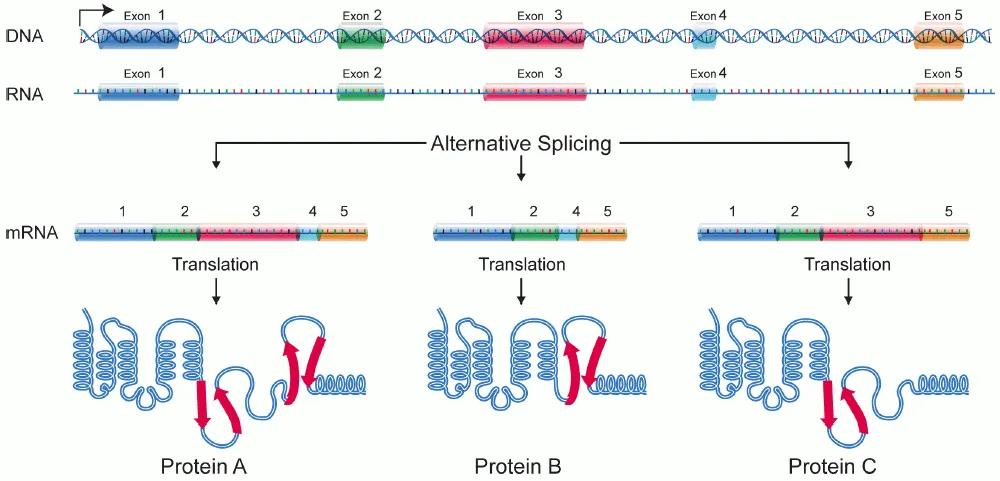
대안적 스플라이싱 과정의 모식도. 하나의 유전자로부터 여러 종류의 mRNA가 만들어지며, 결과적으로 번역되는 단백질의 종류도 다양하다. 출처: 인코덤
흥미롭게도, 많은 유전자에서는 하나의 DNA 서열로부터 여러 가지 다른 mRNA가 만들어질 수 있습니다. 이를 대안적 스플라이싱(alternative splicing)이라고 하는데, 특정 엑손을 포함시키거나 제외시키는 방식으로 조절됩니다. 예를 들어, 어떤 유전자가 5개의 엑손을 가지고 있다면, 1-2-3-5번 엑손만 연결하거나, 1-2-4-5번 엑손을 연결하는 식으로 다양한 조합이 가능합니다.
이러한 대안적 스플라이싱 덕분에 인간 유전체는 약 2만 개의 유전자로부터 10만 개 이상의 서로 다른 단백질을 만들어낼 수 있습니다. 스플라이싱 패턴은 세포 종류나 발달 단계, 질병 상태에 따라 달라지며, 스플라이싱 조절 요소(splicing regulatory element)들과 관련 단백질들에 의해 정교하게 조절됩니다.
폴리아데닐화(polyadenylation)
성숙한 mRNA가 완성되기 위한 마지막 단계 중 하나가 바로 폴리아데닐화(polyadenylation)입니다. 폴리아데닐화는 mRNA의 3' 말단에 아데닌(A) 염기가 연속으로 약 200개 정도 붙는 과정으로, 이렇게 형성된 구조를 폴리-A 꼬리(poly-A tail)라고 합니다.
폴리A 꼬리는 mRNA의 안정성과 번역 효율에 매우 중요한 역할을 합니다. mRNA를 분해하는 효소들로부터 mRNA를 보호하여 반감기를 늘려주며, 또한 폴리A 결합 단백질(PABP)과 결합하여 리보솜에 의한 번역 과정을 촉진시킵니다.
폴리아데닐화가 일어나는 위치는 폴리아데닐화 신호(polyadenylation signal)에 의해 결정됩니다. 대표적으로 AAUAAA라는 서열이 있고, 이 서열 뒤편으로 10-30개 염기 떨어진 곳에서 실제 절단이 일어납니다. 하지만 많은 유전자들이 여러 개의 서로 다른 위치에 폴리아데닐화 신호 서열을 가지고 있어서, 어느 위치의 폴리아데닐화 서열을 사용하느냐에 따라 mRNA의 길이와 안정성이 달라집니다.
이러한 대안적 폴리아데닐화(alternative polyadenylation)는 유전자 발현 조절의 또 다른 중요한 메커니즘으로, 세포 분열, 세포 분화, 스트레스 반응 등 다양한 생물학적 과정에서 활용되고 있습니다. 특히 3' UTR 길이가 달라지면 마이크로RNA(miRNA)의 결합 위치의 수가 변하여 전사 후 조절(post-transcriptional regulation)에도 영향을 미치게 됩니다.
히스톤 변형(histone modifications)

히스톤 변형의 종류. 히스톤을 구성하는 4종류의 단백질 H2A, H2B, H3, H4 꼬리의 특정 아미노산에 특정한 화학적 변형이 일어나며, 이를 히스톤 변형이라고 한다. https://www.cusabio.com/c-20829.html
마지막으로, 염색질 수준에서 유전자 발현을 조절하는 중요한 메커니즘인 히스톤 변형(histone modifications)에 대해 알아보겠습니다.
히스톤 단백질의 아미노산 서열 중 특정 위치에는 다양한 화학적 변형이 일어날 수 있습니다. 대표적으로 라이신(lysine) 잔기에 일어나는 아세틸화(acetylation), 메틸화(methylation), 유비퀴틴화(ubiquitination) 등이 있고, 세린(serine)이나 트레오닌(threonine) 잔기에 일어나는 인산화(phosphorylation) 등이 있습니다.
이러한 히스톤 변형들은 그 종류에 따라 고유한 기능을 가지고 있습니다. 예를 들어, H3K4me3(히스톤 H3의 4번째 라이신의 3-메틸화)는 활발히 전사되는 유전자의 프로모터 부위에서 주로 발견되는 "활성화 마크"입니다. 반면 H3K27me3는 유전자 발현이 억제된 부위에서 발견되는 "억제 마크"입니다. H3K4me1과 H3K27ac는 활성화된 인핸서의 특징적인 마크로 사용됩니다. 주요 히스톤 마크와 연관된 기능을 정리하면 아래와 같습니다.
종류 | 의의 |
H3K4me1 | 주로 인핸서 영역에서 발견되어 인핸서의 활성 상태를 나타냅니다. H3K27ac와 함께 존재할 때는 활성화된 인핸서로 기능하며, H3K27me3와 함께 존재할 때는 특별히 대기 상태(poised)의 인핸서를 나타냅니다. |
H3K4me3 | 활발하게 전사되는 유전자의 프로모터 부위에서 발견됩니다. |
H3K9ac | 활성 유전자의 프로모터 영역에서 발견됩니다. |
H3K9me3 | 이질염색질(heterochromatin)에서 발견되어 유전자 침묵(silencing) 상태를 나타냅니다. |
H3K27ac | 활성 인핸서 및 프로모터에서 발견됩니다. H3K4me1과 함께 활성 인핸서를 나타내는 대표적인 히스톤 마크이며, 전사 활성도가 매우 높고 인핸서 RNA (eRNA) 전사에 필수적입니다. |
H3K27me3 | 발생(development) 조절 유전자의 프로모터 등 넓은 영역에서 나타냅니다. Polycomb repressive complex 2 (PRC2)와 같은 전사를 억제하는 단백질 복합체를 유도하여 유전자 발현을 억제합니다. H3K4me3와 함께 존재할 경우, 발생 과정에서 특정 유전자의 발현 여부를 결정할 수 있는 이중 마크(bivalent mark) 영역을 형성하기도 합니다. |
H3K36me3 | 활성 유전자의 유전자 본체(gene body)에서 발견됩니다. |
이러한 히스톤 변형 패턴은 후성유전학(epigenetics)의 핵심 요소로, DNA 서열의 변화 없이도 유전자 발현을 조절할 수 있게 해줍니다. 중요한 것은 이러한 변형들이 세포 분열을 통해 어느 정도 유지될 수 있다는 점입니다. 이를 통해 세포들이 자신의 정체성을 기억하고 유지할 수 있는 것이죠.
유전체 서열 분석 (게놈 시퀀싱)
유전체 서열분석(게놈 시퀀싱)이란?
인간 유전체는 A, C, G, T를 단위로 하는 약 32억개의 염기쌍으로 구성된다는 것을 앞서 배웠습니다. 사실 32억개의 염기쌍이 가지는 정보의 양은 생각보다는 작은데요, 비유를 한 번 해보겠습니다. A, C, G, T의 4개 글자를 표현하기 위해서는 2개의 비트(bit)가 필요합니다 (00, 01, 10, 11). 즉, 하나의 글자가 2비트의 용량을 차지한다고 본다면, 인간 유전체는 64억 비트를 차지합니다. 64억 비트는 곧 8억 바이트가 되며, 8억 바이트는 대략 800MB(~750MiB)라고 볼 수 있습니다. 인간은 어머니, 아버지로부터 각각 한 세트씩의 유전체 사본을 물려받으므로, 결과적으로 약 1.5GB 정도의 데이터인 셈이네요. 인간이라는 너무도 복잡한 생명체의 설계도가 단지 1.5GB에 불과하다니, 놀랍지 않나요? 과연 이 데이터 안에는 어떤 정보들이 담겨 있는 걸까요?
이 궁금증을 해결하기 위해서는 결국 유전체를 ‘읽어야’ 합니다. 즉, 유전체를 구성하는 A, C, G, T 라는 글자들이 어떤 순서로 나열되어 있는지를 알아내야 하는 것이죠. 이렇게 유전체를 읽는 과정을 염기의 나열(sequence)를 읽는 과정이라 하여, 시퀀싱(sequencing)이라 부릅니다. 시퀀싱 기술의 역사도 무척 재미있는 주제이지만, 본 포스팅에서는 가장 널리 사용되고 있는 차세대 염기서열분석(next-generation sequencing; NGS) 방법 중 몇 가지만 간단히 소개하고 넘어가 보겠습니다.

[참고 - 1세대 시퀀싱]
[참고 - 3세대 시퀀싱]
차세대 염기서열분석법 (next-generation sequencing)
NGS 방법의 핵심은 세포 내에 존재하는 DNA(혹은 RNA)를 무차별적으로 조각낸 다음, 생겨난 임의의 짧은 조각들의 서열을 결정하는 것입니다. 이렇게 먼저 서열 조각들을 읽어낸 뒤, 해당 조각이 유전체(혹은 전사체)의 어느 위치에서 유래되었는지를 역추적함으로써 후속 분석을 진행하게 됩니다. 여기서는 먼저 서열을 읽어내는 기술에는 어떤 것들이 있는지 알아보도록 합시다.
역사적으로 주요했던 NGS 방법론은 아래 4개로 정리해볼 수 있습니다. 현재는 Illumina 사의 방법이 거의 모든 유전체 연구의 표준으로 자리잡았지만, Ion Torrent 시퀀싱은 드물게 임상 진단에서 아직도 사용되고 있는 방법입니다.
•
Roche 454 pyrosequencing
•
개발사: Roche
•
방법 구분: 합성에 의한 시퀀싱(sequencing-by-synthesis; SBS)
•
원리: DNA가 한 염기씩 합성될 때 방출되는 pyrophosphate를 빛 신호로 전환하여 검출합니다. 특정 염기(예: ‘A’)를 넣어줬을 때 빛이 방출되면 해당 위치에는 넣어준 염기와 상보적인 ‘T’가 있다는 것을 의미합니다. 빛이 방출되는 세기를 통해 염기가 몇 개나 연속되는지도 파악할 수 있습니다.
•
특징: 당시로서는 획기적으로 긴 염기서열(약 400~600bp)을 읽을 수 있었지만, 비용이 비싸고 동일 염기 반복 구간(homopolymer) 분석에 오류가 있는 단점이 있었습니다. 현재는 다른 기술에 밀려 시장에서 사라졌습니다.

Metzker (2010)
•
Ion Torrent
◦
개발사: 당시 Life Technologies, 현 Thermo Fisher Scientific 소속
◦
방법 구분: 합성에 의한 시퀀싱(sequencing-by-synthesis; SBS)
◦
원리: 빛(광학 신호) 대신 반도체 칩을 사용합니다. DNA 염기가 가닥에 결합할 때마다 수소이온(H⁺)이 방출되면서 주변의 pH가 미세하게 변하는데, 이 변화를 반도체 센서가 전기 신호로 직접 감지하여 서열을 읽어내는 방식입니다.
◦
특징: 카메라나 레이저 같은 고가의 광학 장비가 필요 없어 기기 가격이 저렴하고 분석 속도가 매우 빠릅니다. 454 기술과 마찬가지로 동일 염기 반복 구간 분석에 약점이 있지만, 빠른 속도 덕분에 임상 현장에서 특정 유전자 패널을 신속하게 검사하는 데 주로 사용되고 있습니다.
•
SOLiD (Sequencing by Oligonucleotide Ligation and Detection)
•
개발사: Applied Biosystems, ABI. 현재는 Thermo Fisher Scientific 소속의 Life Technologies Applied Biosystems Group (Life/APG)
•
방법 구분: 연결에 의한 시퀀싱(sequencing-by-ligation; SBL)
•
원리
◦
DNA 중합효소(polymerase) 대신 DNA 연결효소(ligase)를 사용합니다.
◦
미리 염기 서열을 알고 있는 짧은 형광 표지 조각(프로브, probe)들을 준비합니다.
◦
이 프로브가 분석하려는 DNA 가닥에 정확히 들어맞으면 연결효소에 의해 연결(ligation)됩니다.
◦
이때 프로브에 붙어있던 형광 신호를 읽어 어떤 종류의 프로브가 붙었는지 확인하고, 이를 통해 원래 DNA 서열을 유추합니다.
◦
신호를 읽은 후 형광 물질 부분을 잘라내고 다시 다음 프로브를 붙이는 과정을 반복합니다.
•
특징: 2-base encoding 이라는, 한 번에 한 염기를 읽는 것이 아니라 두 개의 염기(2-base) 정보를 한 번에 읽는 독특한 방식을 사용합니다. 예를 들어, 프로브 하나가 'AG' 조합인지 'AT' 조합인지를 색깔로 구분하는 방식입니다. 이 때문에 시퀀싱 오류와 실제 변이(SNP)를 구분하는 데 매우 정확하다는 장점이 있었습니다.

Metzker (2010)
•
Illumina sequencing
•
개발사: Illumina
•
방법 구분: 합성에 의한 시퀀싱(sequencing-by-synthesis; SBS)
•
원리: 454나 Ion Torrent와 유사하게 ‘합성에 의한 시퀀싱’ 방법이지만, 각 염기(A, C, G, T)에 서로 다른 형광 물질(fluorophore)과 복제 중단 물질(terminator)을 붙여 사용합니다. 한 사이클에 한 염기만 추가되고, 형광 신호를 읽은 뒤 중단 물질을 제거하고 다음 염기를 붙이는 과정을 반복합니다.
•
특징: 압도적인 정확도와 대용량 데이터 생산 능력을 자랑합니다. 분석할 수 있는 염기서열 길이는 상대적으로 짧지만, 수많은 데이터를 생산하여 상호 비교함으로써 정확도를 극도로 높입니다. 거의 모든 유전체 연구의 표준 기술로 자리 잡았습니다.

Metzker (2010)
유전체 서열 분석 방법의 흐름
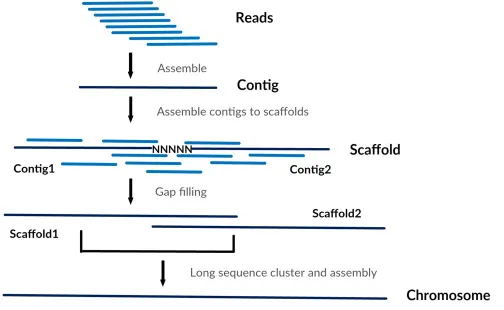
De novo assembly의 흐름. Guo et al. (2017)
위와 같은 시퀀싱 기법을 활용하여 수억 개에 달하는 짧은 시퀀싱 리드(sequencing read)들을 얻어낸 다음, 생물정보학 알고리즘을 활용하여 각 리드에서 겹치는 부분을 찾아내어 조립하는(assemble)과정을 반복하면 개별 염색체 수준의 긴 서열을 얻습니다. 이렇게 아무것도 없는 상태에서 시퀀싱 리드만으로 유전체를 재구성해내는 과정을 드노보 어셈블리(de novo assembly)라 하며, 참고할만한 유전체 정보가 없는 새로운 종에 대한 유전체 서열을 구축하고자 할 때 사용합니다.
이미 알려진 참조유전체(reference genome)이 있는 종에 대한 시퀀싱 실험에서는 각 시퀀싱 리드와 가장 유사한 참조유전체 위치를 찾는 alignment/mapping 과정을 통해 시퀀싱 리드를 좀 더 수월하게 배열할 수 있습니다. 이를 re-sequencing 방법이라 하며, 참조유전체와 아주 약간의 차이를 보이는 개체 수준의 변이를 포착해낼 때 사용하는 방법이 되겠습니다.
기능유전체 분석에서의 시퀀싱의 활용
기능유전체학에서는 앞서 알아본 서열 분석 방법을 유전자 발현, 전사 인자 결합, 염색질 구조 등 다양한 생물학적 현상을 정량화하는 데 이용합니다. 시퀀싱에서 시작하여, 사후 분석이 진행되는 흐름을 단계별로 살펴보면 아래와 같습니다.
1. 참조 유전체/전사체에 매핑
전처리된 리드는 참조 유전체(reference genome) 혹은 참조 전사체(reference transcriptome)에 매핑됩니다. 앞서 배웠듯, 매핑이란 각 리드가 유전체의 어느 위치에서 유래했는지를 역추적하는 과정을 의미합니다. 매핑에는 BWA, Bowtie2, STAR 등의 툴이 사용되며, 매핑 결과는 보통 BAM 또는 SAM 형식의 파일로 저장됩니다.
2. 데이터 분석 및 해석
리드 매핑의 정보를 담고 있는 BAM/SAM 파일은 목적에 따라 다양한 방식으로 분석됩니다:
•
피크 콜링(peak calling): ChIP-seq, ATAC-seq 등에서 신호가 강한 피크(peak) 영역을 식별합니다.
•
발현량 정량화(gene expression quantification): RNA-seq에서 각 유전자의 발현 수준을 계산합니다.
•
차등 분석(differential analysis): 서로 다른 조건 간의 차이를 통계적으로 분석합니다.
•
모티프 분석(motif analysis): DNA/RNA 결합 단백질의 결합 선호도를 나타내는 모티프를 발견합니다.
3. 시각화 및 결과 해석
분석 결과는 유전체 브라우저(IGV, UCSC Genome Browser 등)나 다양한 시각화 도구를 통해 시각화되어 생물학적 의미를 해석하는 데 활용됩니다. 또한, 여러 데이터셋을 통합하여 복잡한 생물학적 현상을 종합적으로 이해하는 데 사용됩니다.
이러한 전체적인 흐름은 모든 유전체 서열 분석 방법에 공통적으로 적용되지만, 구체적인 단계와 방법은 연구 목적(유전자 발현 분석, 염색질 접근성 분석, 단백질-DNA 상호작용 분석 등)에 따라 달라집니다. 또한 기술의 발전에 따라 더 효율적이고 정확한 방법들이 계속해서 개발되고 있습니다.
기능유전체 시퀀싱의 종류
유전자 발현, 염색질 접근성 등 세포의 다양한 기능적인 상태를 포착하기 위해서 다양한 시퀀싱 기법들이 응용되어 왔습니다. 이번 섹션에서는 기능유전체 시퀀싱 기법들의 종류와 원리에 대해 알아봅시다.
유전자 발현 분석을 위한 시퀀싱 방법: RNA-seq, CAGE-seq, PRO-cap
유전자가 실제로 얼마나 활발하게 작동하고 있는지를 알아보기 위해서는 현재 세포 내에 존재하는 메신저 RNA(messenger RNA, mRNA)의 양을 측정하면 됩니다. 세포 내에서 단백질의 합성은 리보솜이 개개의 mRNA를 읽으면서 이루어지기 때문에, 어떤 단백질 정보를 암호화하는 mRNA 양이 많다는 것은 대개 해당 단백질의 양이 많다는 것으로 생각해볼 수 있습니다.
RNA-seq은 시퀀싱을 이용한 전사체 분석의 가장 기본이 되는 분석 범주입니다. 여러 세포가 섞인 샘플의 평균적인 유전자 발현량을 분석하는 벌크(bulk) RNA-seq을 비롯하여, 최근에는 샘플 내 개별 세포 하나하나의 유전자 발현을 분석하는 단일세포(single-cell) RNA-seq 기술이 널리 사용되고 있으며, 더 나아가 조직 내에서 유전자가 발현되는 공간적 위치 정보까지 함께 분석하는 공간전사체학(spatial transcriptomics) 분석 또한 각광받고 있습니다. 여기서는 가장 기본이 되는 bulk RNA-seq 방법에 대해 알아보도록 하겠습니다.
가장 먼저 조직, 세포 등 샘플로부터 전체 RNA(total RNA)를 추출하는 과정을 거쳐야 하며, 분석 목적에 따라서 mRNA만 선택적으로 분리하거나 세포 내 RNA의 대부분을 차지하는 리보솜 RNA (rRNA)를 제거하는 과정이 뒤따릅니다. RNA는 매우 불안정하므로 RNA 자체의 서열을 그대로 읽는 것은 쉽지 않습니다. 따라서 역전사 효소(reverse transcriptase)를 이용하여 RNA를 상보적 DNA (cDNA)로 변환한 다음 시퀀싱을 위한 라이브러리를 구축합니다. 라이브러리 구축에는 cDNA를 적절한 길이로 조각내는 과정, 조각 양 끝에 시퀀싱 기기 내에서의 인식을 위한 짧은 어댑터(adapter) 서열을 붙이는 과정 등이 포함됩니다. 그 다음 NGS 시퀀서를 이용하여 라이브러리 서열을 읽어낸 다음, 얻어낸 수백~수천만개의 리드를 참조유전체(전사체)에 매핑하여 해당 리드가 어느 유전자로부터 유래되었는지 파악한 뒤 각 유전자에 매핑된 리드 수를 세어 유전자 발현량을 정량화합니다.
CAGE-seq (Cap analysis of gene expression by sequencing)은 유전자 발현량을 측정하는 동시에 전사 시작 지점(TSS, transcription start site)의 정확한 위치를 파악하는 시퀀싱 방법입니다. 이 시퀀싱 방법은 전사 시작 지점을 알아내기 위해 (이름에서 알 수 있듯이) mRNA 5’ 말단에 존재하는 캡 구조를 이용합니다 (아래 그림). 5’ 캡에 의해서 mRNA는 엑소뉴클라아제(exonuclease)에 의한 분해로부터 보호되며, 리보솜이 5’ 캡을 인식하여 번역 효율이 올라갑니다. 이러한 기능을 위해 mRNA는 전사되는 동시에 5’ 말단에 7-메틸구아노신이 첨가되는 5’-캐핑(capping)과정을 거칩니다.
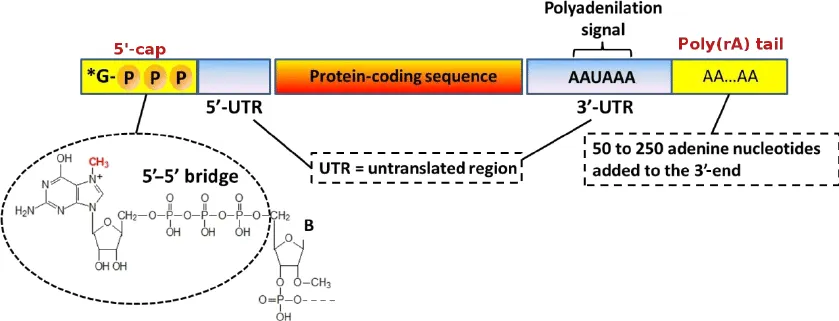
CAGE-seq은 5’ 캡 부분에 비오틴(biotin) 표지 후 스트렙타비딘(streptavidin)이 결합된 magnetic bead를 이용해 5’ 캡이 포함된 RNA 조각만을 선택적으로 선별합니다. 이후 해당 RNA 조각을 주형으로 하는 reverse transcription을 통해 cDNA 라이브러리를 구축하고, 이를 시퀀싱합니다. 결과적으로, 얻어낸 서열들을 참조유전체에 매핑하면 각 유전자의 정확한 전사 시작 부위를 단일 염기 수준에서 확인할 수 있습니다.
PRO-cap (Precision nuclear run-on sequencing coupled with 5’ cap)은, 특정 시점에 특정 위치에 존재하는 RNA polymerase의 위치를 정량하기 위한 PRO-seq (Precision nuclear run-on sequencing) 방법을 TSS 매핑에 응용하는 방법입니다. 전사 중인 RNA를 PRO-seq의 방법과 똑같이 포착해낸 다음, 5’ 말단부터 시퀀싱하게 되면 결과적으로 전사 중인 RNA의 전사 시작 부위를 매핑할 수 있는 원리가 되겠습니다. PRO-seq에 관해서는 아래를 참조해 주세요.

PRO-seq (Precision nuclear run-on sequencing)이란?
염색질 접근성 분석을 위한 시퀀싱 방법: DNase, ATAC-seq
염색질 접근성(chromatin accessibility)은 DNA가 단백질들에 의해 얼마나 쉽게 접근될 수 있는지를 나타내는 지표임을 앞서 배웠습니다. 즉, DNA가 얼마나 노출되어 있느냐에 대한 지표라고 생각하시면 됩니다. 유전자 발현이 일어나기 위해서는 RNA 중합효소와 전사인자들이 DNA에 접근할 수 있어야 하므로, 염색질 접근성은 유전자 발현 조절의 중요한 요소입니다. 이러한 염색질 접근성은 어떻게 정량화해볼 수 있을까요?

Chromatin accessibility를 측정하기 위한 시퀀싱 방법의 종류. Tsompana et al. (2014)
DNase-seq은 가장 전통적인 염색질 접근성 분석 방법 중 하나입니다. 이 방법은 DNase I 효소가 열린 염색질 영역을 우선적으로 자른다는 원리를 이용합니다. 세포에서 추출한 핵에 DNase I을 처리하면, 접근성이 높은 영역(열린 염색질)이 먼저 절단됩니다. 이렇게 생성된 DNA 조각들을 분리하여 시퀀싱하고, DNA 조각들의 위치를 유전체에 매핑해본다면 접근성이 높은 영역을 전체 유전체 수준에서 확인할 수 있겠죠.
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing)은 상대적으로 최근에 개발된 방법으로, Tn5 전이효소(transposase)를 이용합니다. 이 효소는 접근 가능한 염색질 영역에 DNA 어댑터를 삽입하는 두 가지 기능(DNA 절단과 어댑터 삽입)을 동시에 수행합니다. 이 과정을 "태깅(tagging)"이라고 하며, 이후 PCR 증폭과 시퀀싱을 통해 접근성이 높은 영역을 파악할 수 있습니다. ATAC-seq은 DNase-seq에 비해 몇 가지 장점이 있습니다. 첫째, 더 적은 세포(약 500-50,000개)로도 분석이 가능하며, 둘째, 실험 과정이 약 3시간 정도로, 더 단순하고 빠릅니다. 이러한 이유로 ATAC-seq은 현재 염색질 접근성 분석의 표준 방법으로 널리 사용되고 있습니다.
이러한 방법들을 통해 얻은 데이터는 유전체의 어떤 부분이 활성화되어 있는지, 전사인자들이 어디에 결합할 가능성이 높은지 등을 알려주는 중요한 정보를 제공합니다. 특히 프로모터와 인핸서와 같은 조절 영역은 대개 접근성이 높은 영역으로 나타나는 경향이 있습니다.
히스톤 변형 및 전사 인자 결합 분석을 위한 시퀀싱 방법: ChIP-seq
세포 내의 DNA는 많은 단백질과 함께 뒤엉켜 존재합니다. 그 중에서도 히스톤 단백질은 DNA가 감겨 응축될 수 있게 하는 중심 단백질임과 동시에, 사방으로 뻗어 있는 단백질 꼬리에 형성되는 많은 화학적 변형에 의해 유전체 기능의 대표적인 조절자로써 기능합니다. 이러한 화학적 변형들을 히스톤 변형(histone modification) 혹은 히스톤 마크(histone mark)라고 부르며, 기능을 조절하는 히스톤 마크의 조합들을 흔히 히스톤 코드(histone code)라고 부릅니다. 그렇다면 어떤 순간에, 유전체의 특정 부위에 존재하는 히스톤 마크들의 종류와 양은 어떻게 포착할 수 있을까요? (실제로는 히스톤 단백질 상에 형성되는 화학적 변형이지만, 편의상 해당 히스톤에 감겨 있는 유전체 위치에 “히스톤 마크가 형성되었다” 라고 표현하겠습니다.)
특정 히스톤 마크를 인식하여 결합하는 항체와, 서열분석 기법을 함께 활용하면 멋지게 히스톤 마크들의 위치와 양을 파악할 수 있습니다. 가장 먼저, DNA와 히스톤 단백질이 서로 잘 붙어있도록 고정하고, DNA가 적절한 크기로 조각나도록 처리해줍니다 (초음파 처리(sonication)를 할 수도 있고, 효소를 처리할 수도 있습니다). 그 다음, 특정 히스톤 마크를 인식하는 항체를 처리한 뒤 해당 항체를 분리해내면 히스톤 단백질과 함께 결합해있는 DNA 조각들을 얻어낼 수 있겠죠. 이 DNA 조각들을 서열분석하여 유전체에 다시 매핑한다면, 결과적으로 원래 세포에 존재하고 있던 히스톤 마크들의 위치와 양을 알아낼 수 있게 되는 것입니다. 이렇게 항체를 이용하여 DNA-결합 단백질을 분리해내어 해당 단백질이 결합하고 있던 염색질 일부를 획득하는 방법을 염색질 면역침강(chromatin immunoprecipitation, ChIP)법이라 하고, 염색질 DNA 서열 분석까지 결합한 방법을 ChIP-seq이라고 부릅니다. 특히, 특정 히스톤 마크를 인식하는 항체를 이용하는 방법은 히스톤 ChIP-seq (histone ChIP-seq) 방법이라고 합니다.
DNA에 결합한 단백질은 히스톤 단백질 뿐만이 아닙니다. 유전자 발현 조절에 관여하는 전사 인자도 있고, DNA 복제에 관여하는 단백질들도 있고, 심지어 유전체의 3차원적인 구조를 형성하는 뼈대가 되는 단백질들도 있습니다. 앞서 알아본 ChIP-seq 방법을 이용하여 이러한 수많은 단백질에 결합해 있는 DNA 서열을 분석한 뒤 이를 다시 유전체에 매핑한다면, 해당 단백질이 유전체의 어느 위치에 결합하고 있었는지를 역추적해볼 수 있겠죠? 이것이 ChIP-seq 방법을 사용하는 이유입니다.
염색질 접촉 분석을 위한 시퀀싱 방법: Hi-C, micro-C
DNA는 세포 핵 안에서 무작정 뭉쳐있는 것이 아니라, 매우 정교한 3차원 구조를 가지고 있습니다. 마치 실뭉치가 복잡하게 얽혀있는 것처럼 보이지만, 실제로는 특정한 패턴과 규칙에 따라 접혀있는 것이죠. 전의 “인핸서” 파트에서 알아보았듯이 이러한 3차원 구조는 유전자의 발현 조절에 매우 중요한 역할을 하게 됩니다.

Hi-C 방법의 개요. 출처: Wikipedia
Hi-C(High-throughput Chromatin Conformation Capture)는 이러한 염색질의 3차원 구조를 분석하는 대표적인 방법입니다. 기본 원리는 의외로 간단한데요, 세포 안에서 공간적으로 가까이 위치한 DNA 부위들을 화학적으로 연결시킨 후, 제한효소 처리를 통해 적절한 크기로 잘라냅니다. 잘라낸 DNA 조각들을 이어붙이고(ligation), 이어붙인 조각을 paired-end로 한번에 시퀀싱해서 어떤 부위들이 서로 접촉하고 있는지를 알아내는 것입니다.
Paired-end 시퀀싱이란?
앞서 서열분석을 위해서는 가장 먼저 DNA 혹은 RNA를 적절한 크기로 조각내는 것이 필요하다고 했습니다. 이 조각의 서열을 결정하는 데에 있어서는 paired-end 시퀀싱 / single-read 시퀀싱의 크게 두 가지 방식이 있습니다.

Paired-end 시퀀싱은 DNA 또는 RNA 조각의 양쪽 끝을 모두 읽어 염기 서열을 분석하는 방법입니다. 이 방법은 특히 복잡한 유전체/전사체 분석에 유용하게 활용됩니다. 대략의 DNA / RNA 조각 길이를 알고 있을 때, 조각의 양 쪽에서 서열을 결정한 뒤 이 서열이 참조유전체 (혹은 참조전사체)의 어느 부분에서 유래되었는지 역추적하는 방식으로 유전체에 발생한 여러 구조적 변이들을 포착해내거나, 스플라이싱 패턴 등을 정량화할 수 있게 합니다. 반대로 조각의 한쪽 끝만을 읽는 single-read 시퀀싱의 경우에는 paired-end 시퀀싱에 비해 고수준의 정보를 제공하지는 않지만, 비용의 저렴함과 생산되는 데이터의 간결함 때문에 단순한 RNA-seq, small RNA-seq이나 ChIP-seq 등 꼭 paired-end 방법이 필요하지 않은 경우 종종 사용되는 경제성이 있는 방법이라 할 수 있겠습니다.
micro-C는 Hi-C의 개선된 버전으로, 더 높은 해상도로 염색질의 접촉을 분석할 수 있는 방법입니다. Hi-C가 수 킬로베이스(kb) 단위의 해상도를 제공한다면, micro-C는 수백 베이스 단위까지 세밀하게 분석할 수 있어서, 개별 유전자 수준에서의 염색질 상호작용을 더 정확히 파악할 수 있습니다.
Sequence-to-function 모델의 역사
지금까지 AlphaGenome을 이해하기 위한 기본적인 생물정보학 기법들에 대해 알아보았으니, 본격적으로 AlphaGenome 모델에 대해 알아봅시다.

DeepBind. https://www.nature.com/articles/nbt.3300
AlphaGenome은 갑자기 혜성처럼 등장한 모델은 아닙니다. 유전체 서열의 복잡한 문법을 딥러닝이라는 도구로 이해하려는 시도들은 CNN, RNN이 태동하기 시작했던 십여 년 전부터 차곡차곡 그 역사를 쌓아 왔습니다. 제가 기억하는 가장 처음으로 생물정보학 분야에서 현대적인 딥러닝 방법론을 적용하여 유전체 서열을 분석하고자 시도한 사례는 지금으로부터 10년 전인 2015년, DeepBind라는 논문입니다. 너무나 신기했던 논문이라 대학원 신입생 때, 학부 동기들과 함께하는 세미나에서 관련 리뷰 발표를 했던 기억이 새록새록 나네요. 아래에 세미나 자료를 간단히 공유하니, 관심있으신 분은 확인해보셔도 좋을 것 같습니다
자료 보기
다시 본론으로 돌아와서, DeepBind는 한 마디로 DNA 혹은 RNA 서열이 주어졌을 때, 특정 단백질이 그 서열 어딘가에 결합할(binding) 것인지 아닌지를 예측하는 모델입니다. DNA의 경우 주로 TF의 결합을 예측하고, RNA의 경우에는 RNA-binding protein (RBP)의 결합을 예측하게 됩니다. 지금 다시 돌이켜보면 간단한 형태의 CNN 모델에 약간의 AutoML스러운 tuning이 첨가된 연구에 불과하다고 느껴지기도 합니다만, 과장을 조금 보태 AI-(Genome) Biology의 시작이라 할 수 있는, 꽤나 혁신적인 면들을 많이 보여준 마일스톤 논문임에는 틀림없습니다.

BPNet.
그 이후로도 많은 AI 기반 TF-binding 예측 논문들이 있어 왔지만, 개인적으로 좋아하는 연구는 BPNet (2021)과 ChromBPNet (2024)입니다. DeepBind가 단순히 어떤 단백질이 해당 서열에 결합하느냐 아니냐를 분류하는 모델이었다면, BPNet은 ChIP-nexus 기법을 통해 얻은 TF의 binding profile을 (그 이름처럼) 염기쌍(bp) 수준의 해상도로(base-pair resolution) 예측하는 모델입니다. Dilation이 들어간 convolutional layer의 사용으로 입력 서열의 길이를 1kb까지 늘릴 수 있었고 DeepLIFT 와 TF-Modisco 라는 결과 해석을 위한 기법들을 적극적으로 활용해서 “AI가 학습한 결과를 다시 해석하여 insight를 발굴해내는”, AI 기반 생명과학 연구의 패러다임을 보여주고 있습니다. (참고로 DeepLIFT (ICML 2017)은 ‘해석 가능한 인공지능’ 분야에서 꽤나 유명한 기법으로 자리잡았습니다. 보통은 비전이나 언어 분야에서 잘 자리잡은 기술을 가져다 쓰는 것이 일반적인 Bio-AI 분야임을 감안해볼 때, (아마도) 생물학 연구를 위해 처음 개발된 방법이 AI 전반에 큰 파급을 가져다준 몇 안되는 훌륭한 예라고 볼 수 있겠습니다.)


Sequence-to-function 모델에는 염기서열 기반 단백질 결합 예측 모델만 있는 것은 아닙니다. 대표적으로 DNA 서열이 암호화하고 있는 “유전자 발현”에 관한 정보들을 예측하는 모델들이 존재하지요. DNA 서열 그 자체로부터 전사 활성(transcriptional activity)을 예측하는 Xpresso, Expecto와 같은 모델과 더불어, 전사 개시 위치(transcription initiation site) 예측에 특화된 ProCapNet 이라는 모델을 예로 들 수 있겠습니다. 더 나아가서, SpliceAI와 Pangolin과 같이 전사 이후의 mRNA 스플라이싱을 예측하는 모델들 또한 존재합니다. (SpliceAI 논문은 2019년에 Cell에 발표된 비교적 오래된 논문이지만, 잘 학습된 Genomic AI 모델을 이용하여 생물학적인 통찰을 도출하는 과정이나, AI 예측의 해석 결과를 유전체 변이의 임상적인 의의와 연관짓는 분석 흐름이 잘 짜여진 논문으로, 한 번 읽어보시기를 추천드립니다.)
유전체는 세포핵 내에서 매우 복잡한 3차원 구조를 형성하고 있습니다. 유전체끼리 맞닿아 고리를 이루고 있는 부분도 있고, DNA 가닥이 느슨하게 풀려 있는 부분도 존재하며, 핵막 근처에 존재하여 단단하게 응축되어 있는 부분들도 있습니다. 그런데 놀랍게도, 유전체의 3차원 구조 정보 또한 유전체 서열로부터 예측이 가능합니다. (아마도 3D genome의 구조를 시각적으로 확인하는 실험 기법인 Optical Reconstruction of Chromatin Architecture(ORCA)에서 이름을 따온 것으로 보이는) Orca 모델은 이것이 가능함을 보여준 대표적인 예입니다.
AlphaGenome, 약 10년에 걸친 모델 개발사
앞선 모델들의 공통점은 단순합니다. 바로 DNA 서열을 입력으로 받아 유전체의 기능적인 상태를 예측한다는 점이죠. 자연스럽게 “DNA 서열을 입력으로 받아 여러 모달리티(modality), 혹은 오믹스(omics) 차원의 활성도를 동시에 예측하는 멀티모달(multi-modal) 예측 모델을 만들어보면 어떨까?” 라는 아이디어를 떠올릴 수 있고, 실제로 Basset (2016), Basenji (2018, 2020), Enformer (2021), Borzoi (2025)와 같은 모델들이 그러한 아이디어를 바탕으로 개발되어 왔습니다. 흥미롭게도, Basset부터 Borzoi까지 이어지는 모델 시리즈들은 모두 구글의 자회사 Calico Life Sciences와 DeepMind 연구진들이 개발한 모델들이고, 어떻게 보면 약 10년에 걸쳐 개발되어 온 AlphaGenome의 전신 모델들이라고 볼 수도 있겠습니다.
(한가지 더, basset, basenji, borzoi는 모두 하운드 계열 견종의 이름들입니다. 아래 그림을 보시면… 모델이 발전함에 따라 점점 길쭉해지는군요 )
)


사실 Basset은 DNA 서열로부터 DNase-seq으로 얻은 DNA accessibility만을 예측하기 때문에, 엄밀하게는 멀티모달 예측 모델이라고 볼 수는 없겠습니다만, 여기서는 모델 개발 흐름을 알아보기 위해 함께 살펴보겠습니다.
아래 그림은 Basset, Basenji, 그리고 Borzoi 모델 구조를 나타냅니다. 이 그림을 보면서, 모델 구조 측면에서의 발전을 함께 살펴보도록 하겠습니다.
Basset은 2016년에 개발된 모델인만큼, 가장 기본적인 CNN 기반 예측 모델의 형태를 띠고 있습니다. Basenji는 이보다 약간 발전된 형태로, dilated convolution 연산을 사용하고 있네요. 하드웨어의 발전과 더불어, 이 연산의 사용으로, 모델의 입력으로 제공되는 DNA 서열의 길이(window size)가 아주 비약적으로 증가했음을 알 수 있습니다 (0.6kb → 131kb).
Borzoi 모델은 약간 더 고도화된 형태인, downsampling → transformer blocks (attention blocks) → upsampling 으로 이어지는 모델 구조를 가지고 있고, 또한 downsampling layer와 upsampling layer 사이에 U-Net 스타일의 connection이 존재합니다. (Dilated) Convolution 연산만으로는 효율적으로 포착하기 어려운 long-range interaction들을 일련의 downsampling을 거쳐 “저해상화”된 feature space 상에서의 attention으로 포착해내고, downsampling 경로 상의 feature들을 U-Net skip connection을 통해 직접 더해 줌으로써 여러 층위의 해상도에 걸친 feature들을 예측에 활용할 수 있게 합니다. (참고로, 이 구조는 큰 틀에서 AlphaGenome 모델 구조와 아주 유사합니다!)

Basset 모델 구조. 0.6kb window의 DNA 서열을 입력으로 받는다.

Basenji 모델 구조. 131kb window의 DNA 서열을 입력으로 받는다.

Borzoi 모델 구조. 524kb window의 DNA 서열을 입력으로 받는다.
AlphaGenome, 무엇을 예측하는 모델인가?
AlphaGenome의 전신 모델들에 대해 간단히 알아보았으니, 이제 AlphaGenome이 어떤 모델인지 본격적으로 알아보도록 합시다.
세포의 유전체가 어떻게 활용되고 있는지, 그 기능적인 상태는 어떻게 포착할 수 있을까요? 세포핵 내에 DNA가 어떤 형태로 존재하고, 어떤 일을 하고 있는지 사진이나 동영상이라도 찍을 수 있으면 좋을 텐데, 쉬운 일은 아닐 겁니다. 이렇게 유전체 상태를 우리 눈으로 직접 확인할 수는 없기에, 생물정보학 분야에서는 다양한 시퀀싱 기법에 기반하여 간접적인 방법으로 유전체의 기능적인 상태를 포착하는 기법이 활발히 개발되어 오고 있습니다. (다양한 시퀀싱 기법에 대한 개요는 앞 섹션을 참고해 주세요.)
시퀀싱을 통해 어떻게 유전체의 활성 상태를 포착할 수 있는지는 앞선 섹션에서 다루어 보았는데요, 각각의 방법마다 고유의 시료 처리 방법과 분석 방법이 있지만 핵심 원리는 모두 같습니다.
유전체의 어떤 지역 에서 포착하고자 하는 기능적인 활성도)가 있다고 합시다. 예를 들어, 은 유전자 발현량, 특정 전사 인자가 결합되었는지 아닌지의 여부, 염색질이 얼마나 열려 있는지(염색질 접근성) 등이 될 수 있습니다.
이 때, 세포 시료에 모종의 처리를 하여 시퀀싱했을 때 “이 클수록 유전체 지역 에서 유래된 서열 조각들이 많아지게” 할 수 있다고 가정해 봅시다.
그러면 시퀀싱 데이터(기계로 읽어낸 수많은 서열 조각들)를 다시 참조유전체에 매핑했을 때 유전체 지역 에 매핑된 서열 조각의 양은 곧 해당 유전체 지역의 기능적인 활성도를 나타내는 값이라고 생각할 수 있습니다. (”참조유전체 매핑”의 의미에 대해서는 앞 섹션을 참고해 주세요.)
AlphaGenome 논문 결과 데이터를 이해하기 위해서는 위의 원리를 이해하는 것이 필수적이므로, 몇 가지 예를 들어 좀 더 쉽게 이해해 봅시다.
유전체 지역 이 어떤 유전자 X이고, 측정하고자 하는 기능적인 활성도 값이 유전자 발현량이라면 어떤 시퀀싱을 해야 할까요?
•
대표적으로 RNA 시퀀싱(RNA-seq) 이라는 방법을 이용하면 됩니다. 간단히 설명하자면 RNA-seq은, DNA를 읽는 대신 세포 내에 존재하는 RNA 서열 조각들을 읽어 내는 방법입니다. 따라서 유전자 발현량이 큰 유전자의 경우 해당 유전자에서 유래된 서열 조각들이 많아질 테고, 결과적으로 위의 원리를 만족하게 되는 것이죠!
주로 조절 부위(프로모터/인핸서)를 포함하는 임의의 유전체 지역 에 대해, 측정하고자 하는 기능적인 활성도 값이 특정 전사 인자의 결합이라면 어떤 시퀀싱을 해야 할까요?
•
전사 인자 염색질 면역 침전 시퀀싱(TF ChIP-seq) 방법을 이용하면 됩니다. 간단히 설명하자면 이 방법은 “TF가 결합하여 붙잡고 있는” 서열을 TF가 더이상 놓아버리지 못하도록 단단히 결합시킨 후, TF에 특이적으로 결합하는 항체를 이용하여 시료에서 TF를 분리한 다음 TF가 붙잡고 있던 서열을 읽어 내는 방법입니다. 따라서 많은 세포에서 TF가 공통적으로 결합해있던 유전체 지역의 경우 해당 지역을 “붙잡고 있던” TF가 많을 테고, 결과적으로 위의 원리를 만족하게 됩니다.
•
참고로 특정 히스톤 변형(히스톤 마크)의 양을 정량화할 때도 히스톤 ChIP-seq(histone ChIP-seq)이라는 유사한 방법을 사용합니다. 특정 히스톤 마크를 보유한 히스톤 단백질을 특이적으로 분리하여 “히스톤을 휘감고 있던” 서열을 읽어 내는 원리가 되겠습니다.
임의의 유전체 지역 에 대해, 측정하고자 하는 기능적인 활성도 값이 염색질 접근성이라면 어떤 시퀀싱을 해야 할까요? 즉, 염색질이 얼마나 열려 있는지 측정하고 싶다면 어떻게 해야 할까요?
•
DNase-seq 혹은 ATAC-seq 등의 방법을 이용하면 됩니다. 이 방법들의 공통적인 원리는 바로 열린 염색질 부위에만 접근하여 작동하는 효소를 처리해준다는 것입니다. DNase-seq의 경우는 열린 염색질 부위에서 서열을 잘라내는 DNase를 처리하고, ATAC-seq의 경우에는 Tn5 transposase 효소를 처리하여 서열을 잘라내면서 서열분석에 사용되는 어댑터 서열로 태깅(tagging)하는 방법입니다. 결과적으로 이러한 효소들이 잘 처리되는 부분(즉, 열린 염색질 부위)에서 더 많은 서열 조각들이 유래하게 되고, 역시 위의 원리를 만족하게 됩니다.
마지막으로, 앞서 배운 것들을 조금 응용해봅시다. 임의의 두 유전체 지역 및 에 대해서, 측정하고자 하는 기능적인 활성도 값이 두 지역의 공간적 접촉이라면 어떤 시퀀싱을 해야 할까요?
•
Hi-C와 같은 방법을 이용합니다. 간단히 설명하면, 먼저 세포 내의 유전체 3차원 구조가 그대로 유지되도록, 즉 서로 붙어 있는 두 유전체 지역(, )이 떨어지지 않도록 cross-linking을 수행하고, 그 상태로 제한효소를 처리하여 DNA를 잘라냅니다. 공간적으로 붙어 있던 두 지역 , 는 아직도 붙어 있다는 사실이 중요합니다. 이제 서로 붙어 있는 두 DNA 조각들을 서로 이어붙이고(ligation), 서열분석을 수행합니다.
•
이렇게 읽은 서열 조각의 양쪽 끝을 읽었을 때의 결과를 상상해 볼까요? 한쪽 끝은 에서 왔고, 다른쪽은 에서 왔을 겁니다. 결과적으로 이러한 서열 조각의 쌍을 참조유전체에 다시 매핑한다면, 원래 세포에서 공간적으로 접촉하고 있던 유전체 부위들을 역추적할 수 있게 됩니다.
위 모든 서열분석 방법의 결과로 우리는 각 유전체 위치에 대해 매핑된 서열 조각의 양을 얻게 되고, 이를 유전체의 기능적인 활성도와 동치로 보고 분석을 수행합니다. 이 때, 각 유전체 위치에 대해 매핑된 서열 조각의 양을 시각화한다면 아래와 같이 genomic track이라 부르는, 좌우로 길다란 그래프를 얻게 됩니다.

Genomic track의 예시들.
모델 입출력: AlphaGenome은 서열로부터 다양한 genomic track을 예측한다
Genomic track에 대한 이해를 갖추었으니, 이제 우리는 AlphaGenome이 무엇을 예측하는 인공지능 모델인지를 이해해볼 수 있습니다.

Fig.1a - AlphaGenome 모델의 입출력 구조 개요.
위 그림에서 AlphaGenome 모델의 작동 방식을 간단히 확인할 수 있습니다. 예상할 수 있듯이, 모델의 입력으로는 1Mb 길이의 DNA 서열이 활용되며, 이렇게 긴 DNA 서열은 짧은 길이의 서열 조각들로 분할되어 개별 device (GPU) 상에 탑재되어 있는 AlphaGenome 모델을 거치게 됩니다. 그림에서 Encoders-Transformers-Decoders 로 표현되어 있는 부분이 바로 AlphaGenome 모델을 나타내는 부분입니다.
모델의 출력으로는 입력으로 제공된 1Mb 서열 윈도우에 대한 genomic track들이 예측되어 나오게 됩니다. 유전자 발현을 포착하는 RNA-seq, CAGE, PRO-cap 부터, 유전체 접촉을 나타내는 contact map까지 굉장히 다양한 모달리티를 예측하고 있음을 알 수 있습니다. 그림의 오른쪽에는 학습에 사용한 genomic track의 개수가 나타나 있고, 각 모달리티 별로 몇 염기쌍(base-pair; bp) 해상도로 예측을 수행하도록 설정되어 있는지가 나타나 있습니다. 대부분의 경우 단일 염기 수준에서 genomic track의 값을 예측하지만, genomic track을 획득할 때 사용한 시퀀싱 방법 자체의 특성 상 1bp 수준의 해상도가 큰 의미가 없는 경우(ChIP-seq과 Hi-C 실험)에는 1bp 해상도를 고집하고 있지는 않는 모습입니다.
모델 성능: track prediction / variant effect prediction에서, 대부분의 최신 모델보다 나은 성능을 보인다
AlphaGenome 모델의 성능을 확인하기 위해서 생각해볼 수 있는 가장 직관적인 평가 방법은 당연히 genomic track에 대한 정량적인 예측이 잘 이루어지고 있는지를 측정하는 것입니다. 아래 그림은 각 모달리티별로 어떤 평가 metric을 사용했으며, 해당 모달리티에 특화된 기존 모델과 비교했을 때 상대적으로 얼마나 성능 향상이 있는지를 보여주고 있습니다.

Fig. 1d - Track prediction 성능 평가 metric 및 기존 모델과의 성능 비교
Metric 항목을 보면 “여기가 splice site인가”를 예측해야 하는 splice site classification 문제를 제외하고는 모두 Pearson 및 Spearman 상관관계를 평가 metric으로 사용하고 있네요. Track의 높은 곳은 높게, 낮은 곳은 낮게 잘 예측하고 있는지를 평가하고 있다고 보면 되겠습니다.
우측 Comparison 항목에서는 AlphaGenome과 같이 서열 기반으로 genomic track을 예측하는 기존 모델들과 성능을 비교하고 있습니다. 그래프는 기존 모델 대비 AlphaGenome의 성능 향상 정도를 보여주고 있는데, 대부분의 task에서 AlphaGenome이 성능 향상을 보이고 있음을 알 수 있습니다.
한편, AlphaGenome의 활용은 단순히 정해진 서열로부터 다양한 genomic track을 예측할 수 있는 것에 그치지 않습니다. 많은 질병은 유전체 상의 잘못된 정보, 즉 유전 변이에 의해 발생합니다. 이 변이는 부모님의 유전체로부터 물려받은 변이일 수도 있고, 유전체 복제 오류나 암 유발인자에 의한 염기의 화학적 변형 등 외부 요인에 의해 발생한 변이일 수도 있습니다. 서열이 주어졌을 때 해당 유전체의 기능적 활성을 예측하도록 학습된 AlphaGenome은 과연 “변이된 서열”이 주어졌을 때 “유전체의 기능적 이상”을 잘 예측할 수 있을까요?

Fig. 1e - Variant effect prediction 성능 평가 metric 및 기존 모델과의 성능 비교
위 그림의 Modality 항목에서는 각 variant가 splicing, RNA expression, DNA accessibility, TF binding에 미치는 영향을 모델이 잘 예측하고 있는지를 평가하고 있음을 알 수 있고, Evaluation 항목은 개별 벤치마크 세팅의 이름을 보여주고 있습니다. 역시 Comparison 항목에서는 각각의 task에서 가장 높은 성능을 보이고 있던 기존의 모델 대비 대체로 성능 향상이 있음을 보여주고 있네요.