Sequenza
Sequenza is a tool to analyze genomic sequencing data from paired normal-tumor samples, including cellularity and ploidy estimation; mutation and copy number (allele-specific and total copy number) detection, quantification and visualization.
Documentation
Paper
설치
Python 기반의 preprocessing sequenza-utils → R 기반의 plot 으로 분석이 진행된다. 따라서 Python 설치, R 설치 두번의 설치가 필요하다.
mamba install -c bioconda sequenza-utils r-sequenza
Shell
복사
Quickstart
1.
Process a FASTA file to produce a GC Wiggle track file.
sequenza-utils gc_wiggle -w 50 --fasta hg38.fa -o hg38.gc50Base.wig.gz
Shell
복사
2. Process BAM and Wiggle files to produce a seqz file.
sequenza-utils bam2seqz -n normal.bam -t tumor.bam --fasta hg38.fa \
-gc hg38.gc50Base.wig.gz -o out.seqz.gz
Shell
복사
3. Post-process by binning the original seqz file.
sequenza-utils seqz_binning --seqz out.seqz.gz -w 50 -o out small.seqz.gz
Shell
복사
Analysis in R (run_sequenza.R)
library(argparse)
library(sequenza)
parser = ArgumentParser()
parser$add_argument('-i', '--input', required=TRUE) # mysample.small.seqz.gz
parser$add_argument('-n', '--name', required=TRUE) # mysample
parser$add_argument('-o', '--outdir', required=TRUE) # result/mysample
args = parser$parse_args()
extracted = sequenza.extract(args$input)
CP = sequenza.fit(extracted)
sequenza.results(
sequenza.extract=extracted,
cp.table=CP,
sample.id=args$name',
out.dir=args$outdir'
)
R
복사
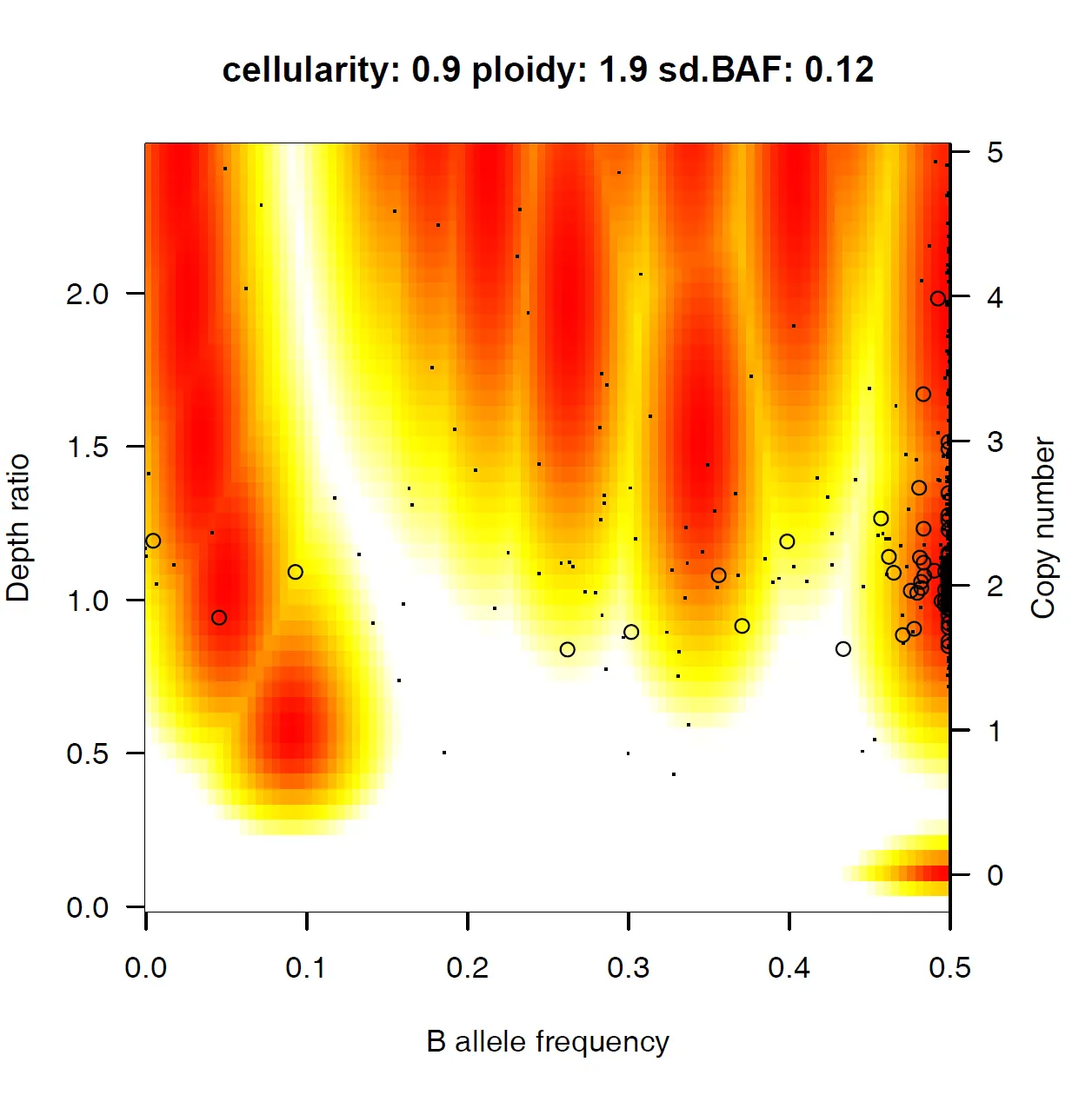
결과들
txt files
•
{mysample}_alterative_solutions.txt
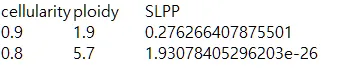
•
{mysample}_confints_CP.txt
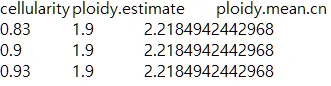
•
{mysample}_mutations.txt
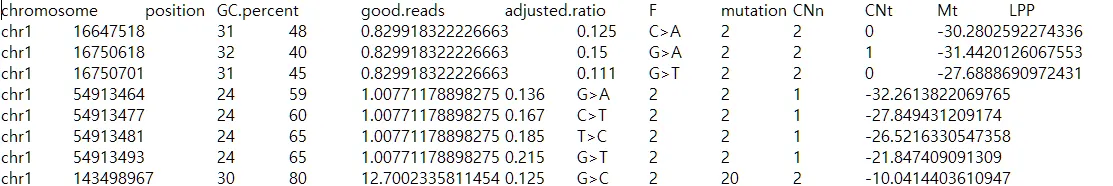
•
{mysample}_segments.txt

pdf files
•
{mysample}_alternative_fit.pdf
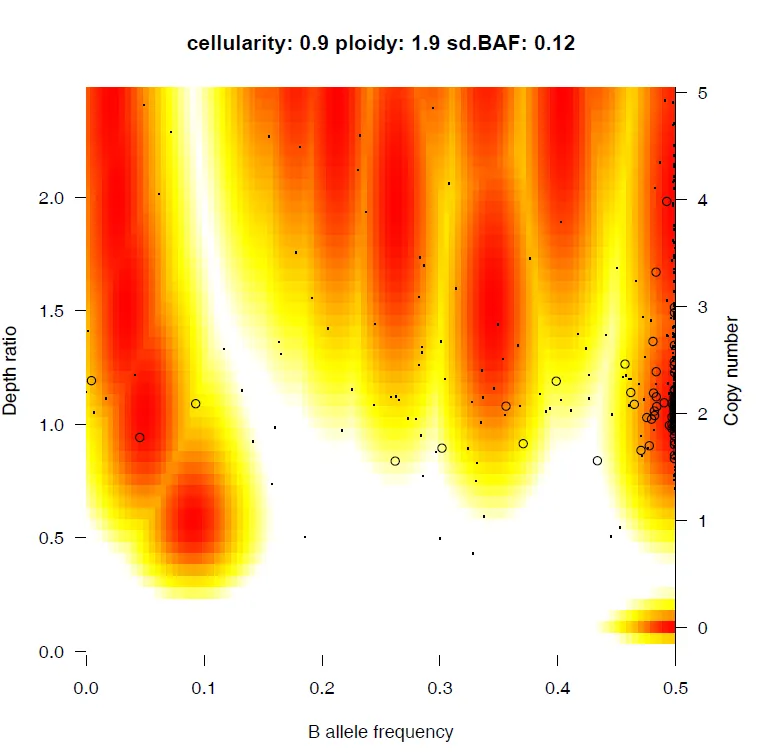
•
{mysample}_chromosome_depths.pdf
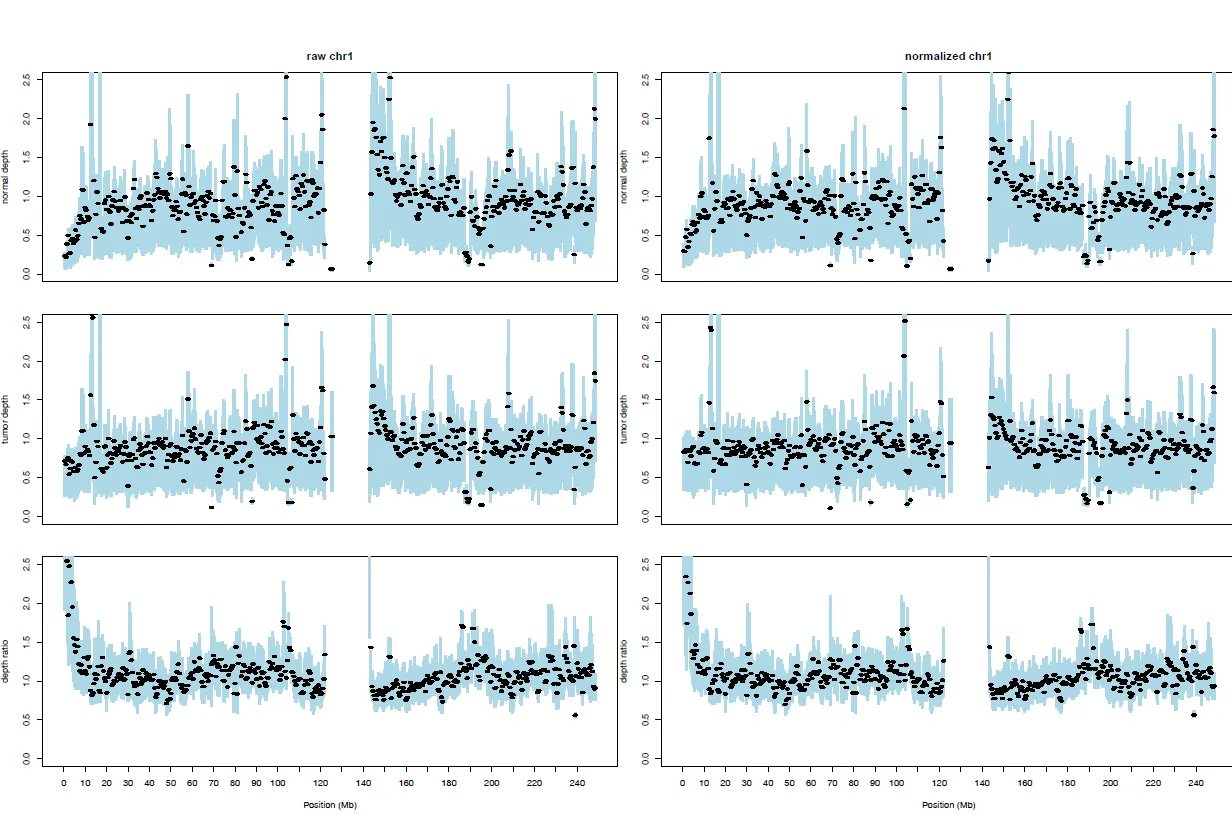
•
{mysample}_chromosome_view.pdf
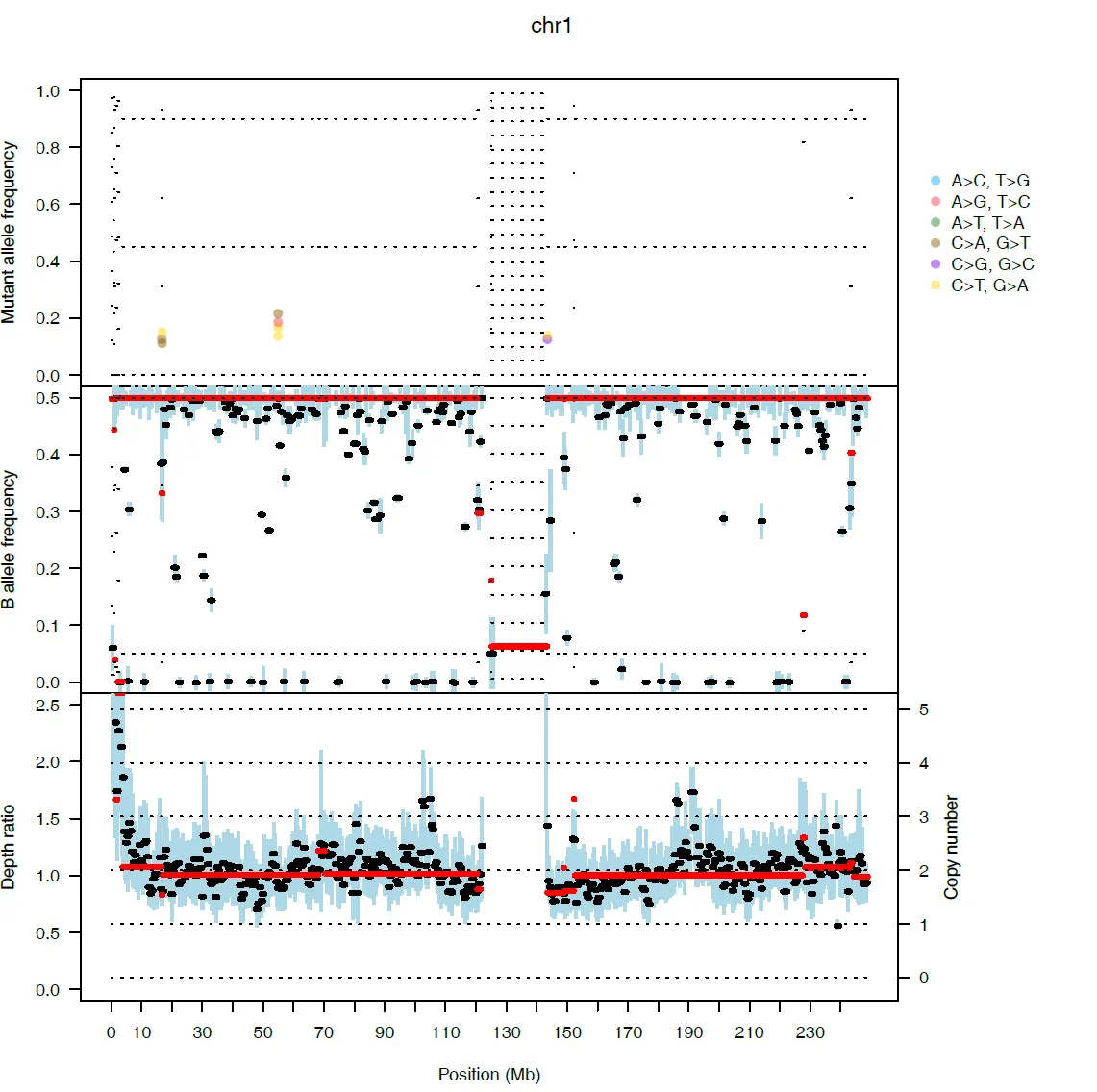
•
{mysample}_CN_bars.pdf
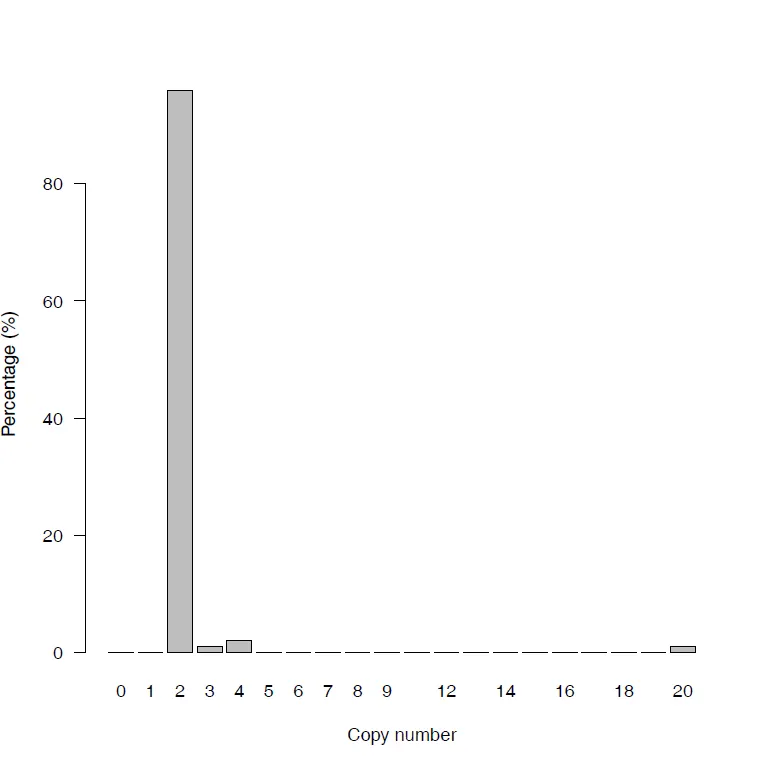
•
{mysample}_CP_contours.pdf
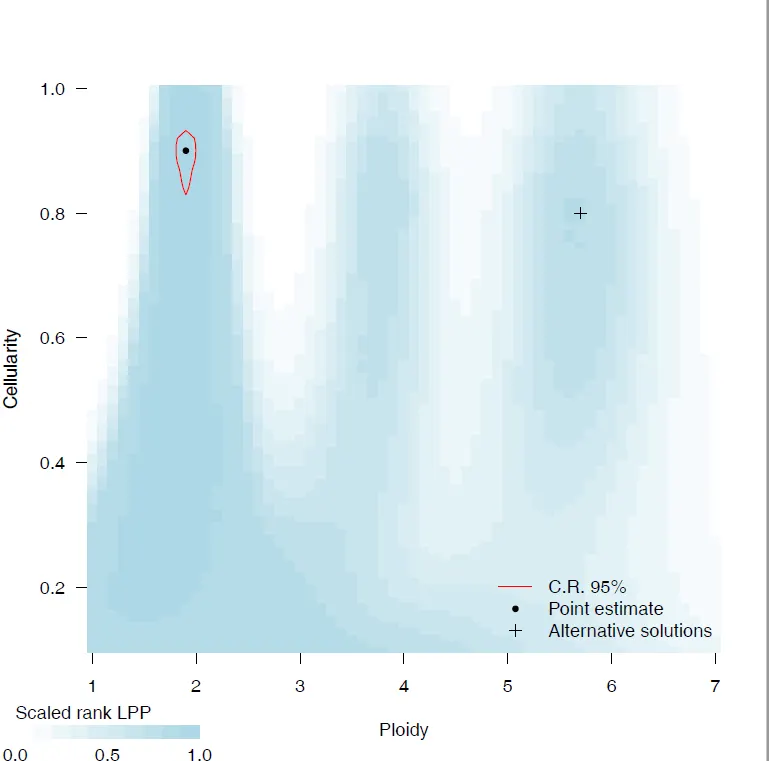
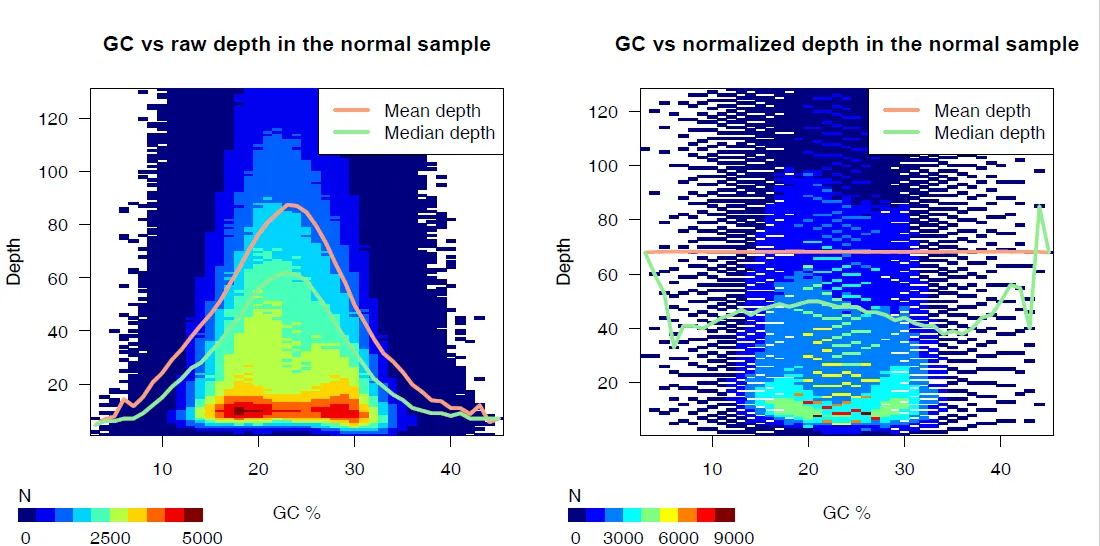
•
{mysample}_gc_plots.pdf
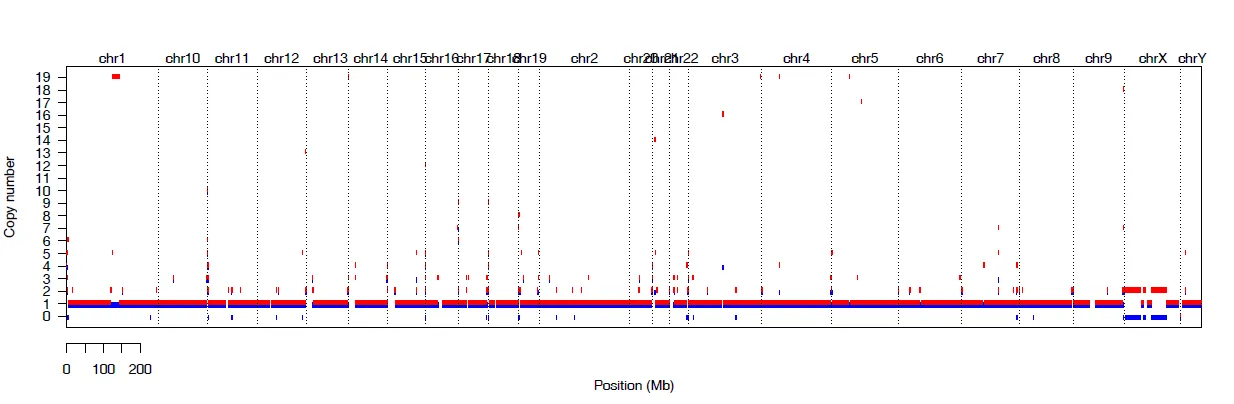
•
{mysample}_genome_view.pdf
•
{mysample}_model_fit.pdf