

AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model
Track prediction 성능 평가
앞서 AlphaGenome이 state-of-the-art 수준의 track prediction 성능을 보여주는 것을 Pearson/Spearman 상관관계 수준에서 확인해 보았습니다. Figure 2에서는 이렇게 우월한 track prediction 성능을 나타내는 자세한 예시들을 선보이고 있습니다.
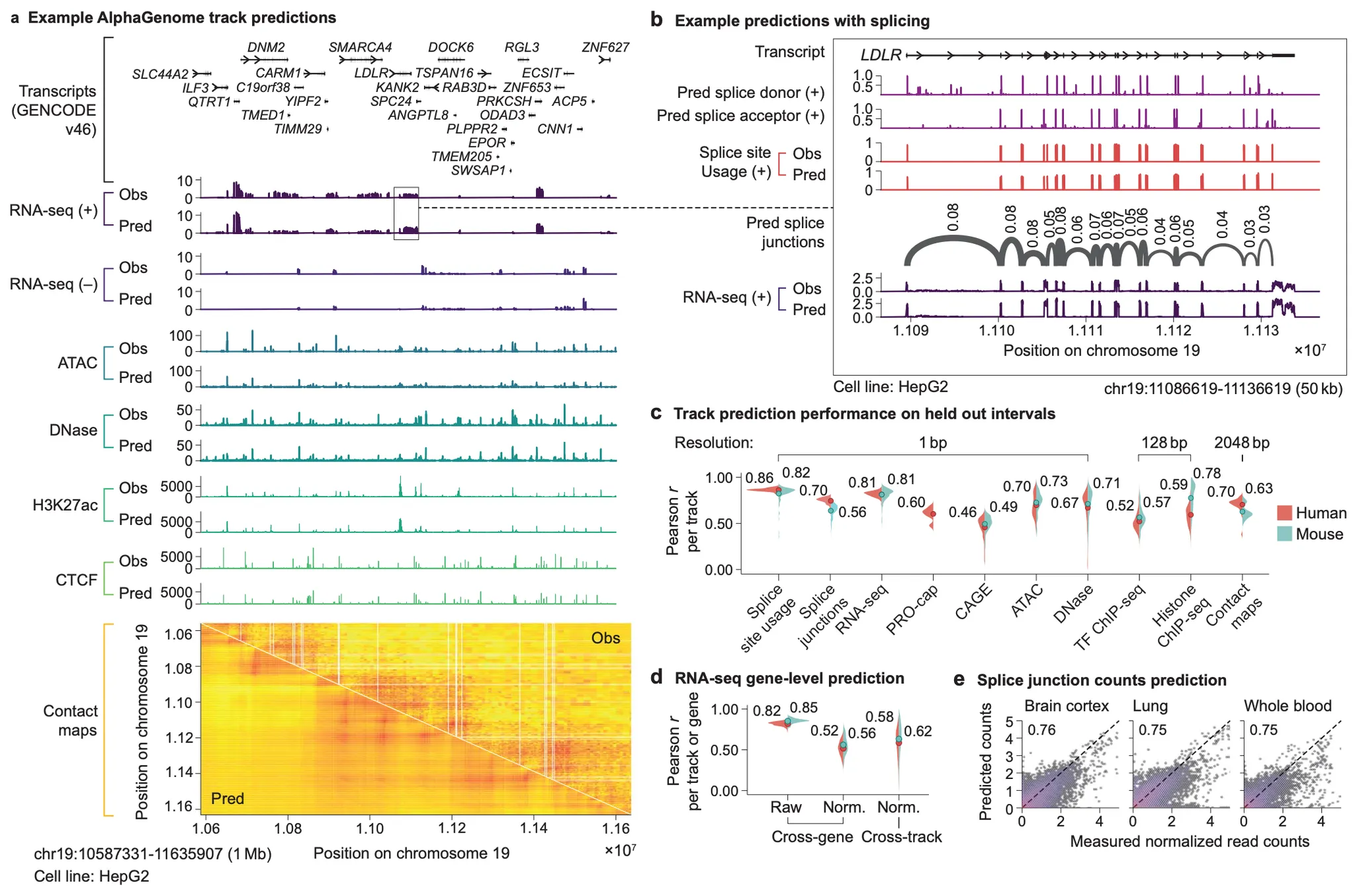
Fig. 2 - AlphaGenome track prediction 결과 예시.
Fig 2a는 chr19:10587331-11635907 (1,048,576bp) 지역 ~1Mbp window에 대한 AlphaGenome 예측을 보여주고 있는데요, 아래와 같이 관찰들을 정리해볼 수 있겠습니다. (참고로 이 figure를 그리는 데 사용한 AlphaGenome 모델의 학습 데이터에는 이 지역이 포함되지 않았다고 합니다.)
•
(Fig. 2a) AlphaGenome은 cell-line 특이적인 track prediction이 가능하다.
◦
HepG2 cell line의 해당 지역 genomic track 예측을 보여주고 있습니다.
•
(Fig. 2a) 다양한 모달리티에 대한 AlphaGenome의 예측은 실제 관찰 결과와 많은 부분 좋은 상관관계를 보인다.
◦
RNA-seq 부터 CTCF까지, 위의 Obs(observed) track과 아래의 Pred(prediction) track이 좋은 일치도를 보입니다.
◦
Contact map 항목에 대해서도 TAD 수준에서 chromatin contact의 경향이 잘 예측되고 있음을 알 수 있습니다.
•
(Fig. 2a) RNA-seq track (즉, 유전자 발현량)을 strand-specific 하게 예측한다.
◦
즉, (+)와 (-) strand 중 어느 가닥을 주형으로 유전자가 발현될 것인지 예측이 가능합니다.
•
(Fig. 2b) RNA-seq track은 exon-특이적으로 coverage를 보인다.
◦
즉, exon-intron 구분 능력이 내재하고 있다고 볼 수 있겠습니다.
•
(Fig. 2b) Splice-site 예측이 가능하며, 정량적인 splice-site usage 예측 또한 가능하다
◦
위에서 알 수 있듯이, exon-intron 구분 능력이 직접적으로 pred splice donor/acceptor 항목에서 나타나고 있으며, 더 나아가서 어떤 splice donor-acceptor pair가 얼마나 선호될지, splice junction usage 예측 또한 가능함을 보여주고 있습니다.
•
(Fig. 2c) Held-out interval에 대해 예측-관측 값 사이에 좋은 상관관계를 보인다
◦
앞선 Fig. 1d 결과와 크게 다르지 않은 결과인 듯 합니다.
•
(Fig. 2d) RNA-seq 결과로부터 유전자 수준 발현량을 재구성한 성능도 어느 정도 좋은 상관관계를 보인다.
참고) 그림 내 각 항목의 의미
◦
단, 가장 왼쪽의 좋은 상관관계는 어느 정도 유전자 발현량의 스케일이 유전자별로 다르기 때문에 (예를 들어 ACTB, GAPDH 등의 housekeeping gene들의 유전자 발현량은 다양한 세포 종류에서 대부분 상위권에 속하는 편입니다) 꽤 쉽게 달성할 수 있는 상관관계 수준이라고 볼 수 있습니다.
◦
다양한 tissue-/cell-type을 고려하여 유전자 발현량을 normalize한 다음 상관관계를 측정 시에는 상대적으로 더 낮은 상관관계를 보이는 것으로 보아, 보다 정밀한 세포 특이적(cell-type specific) 유전자 발현량 예측을 위해서는 조금 더 개선이 필요해 보입니다.
•
(Fig. 2e) Splice junction read count 예측 또한 어느 정도 좋은 상관관계를 보인다.
Variant effect prediction 성능 평가
스플라이싱(splicing) 변화 예측
유전 변이가 질병을 일으키는 원인은 다양합니다. 아미노산을 암호화하고 있는 염기에 변이가 생겨 단백질을 구성하는 아미노산이 달라지게 되면, 종종 단백질의 기능이 망가지고 이에 따라 질병이 생기곤 합니다. 한편, 아미노산을 암호화하는 위치는 아니지만 mRNA 스플라이싱 과정에 중요한 위치에 생기는 변이들도 존재하며, 적지 않은 질병이 이렇게 스플라이싱을 망가뜨리는 변이에 의해서 발생합니다. 스플라이싱에 관여하는 염기서열 모티프(motif)들은 비교적 잘 알려져 있기 때문에, 저자들은 가장 먼저 AlphaGenome을 통해 특정 변이가 스플라이싱에 미치는 영향을 예측할 수 있을지 확인해봅니다.

스플라이싱을 결정하는 서열 모티프
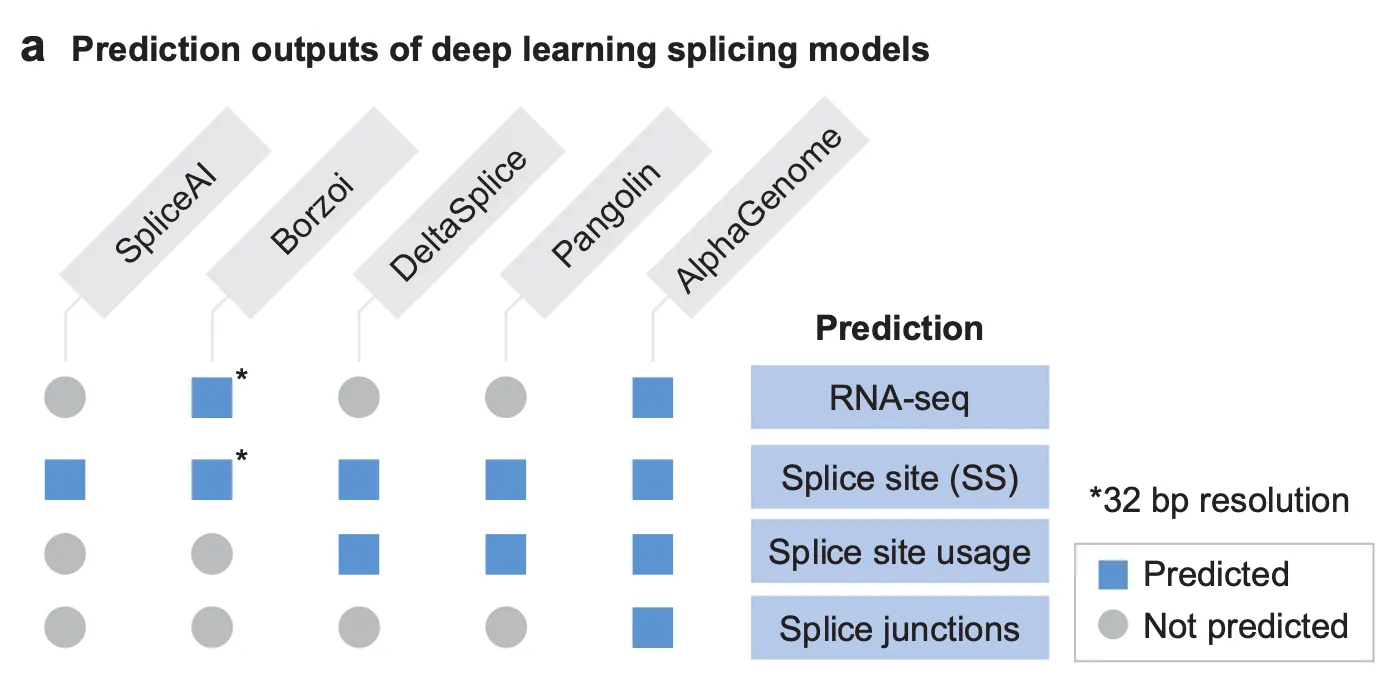
Fig. 3a - AlphaGenome 및 관련 모델들이 예측 가능한 스플라이싱 관련 모달리티 정리.
AlphaGenome은 스플라이싱에 관하여 무엇을 예측할 수 있을까요? 저자들은 스플라이싱의 결과는 3가지 수준에서 예측될 수 있다고 제시하며, RNA-seq track 예측과 더불어 이 3가지 요소를 동시에 예측할 수 있는 모델은 AlphaGenome이 최초라는 점을 강조합니다.
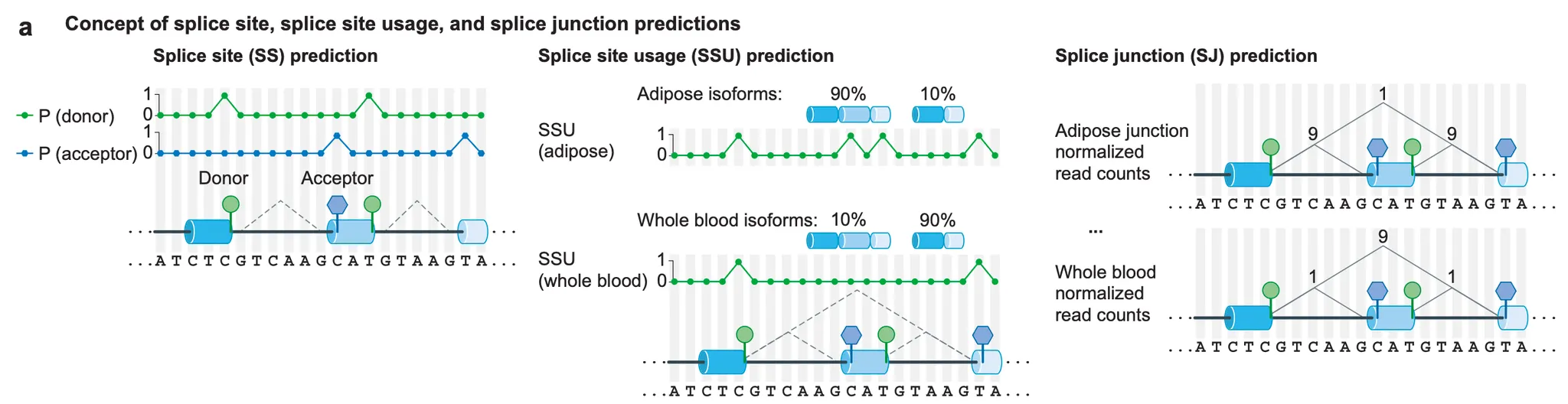
Extended Data Fig. 2a - Splice site prediction, splice site usage prediction 및 splice junction prediction의 모식도.
1.
특정 염기가 splice donor 혹은 splice acceptor로서 기능할 확률을 예측하는 것 (= splice site prediction)
2.
여러 개의 가능한 splice donor 혹은 splice acceptor가 있을 때, 어떤 것이 어떤 비율로 선택되어 스플라이싱이 일어날지 예측하는 것 (= splice site usage prediction)
3.
어떤 exon 조합으로 mRNA가 만들어질지 예측하는 것 (= splice junction prediction)
그 다음, 저자들은 AlphaGenome을 활용하여 스플라이싱 관련 변이들의 효과를 제대로 예측해낼 수 있다는 예시를 몇 가지 보여줍니다.
예시 1) 변이에 의한 exon skipping 예측
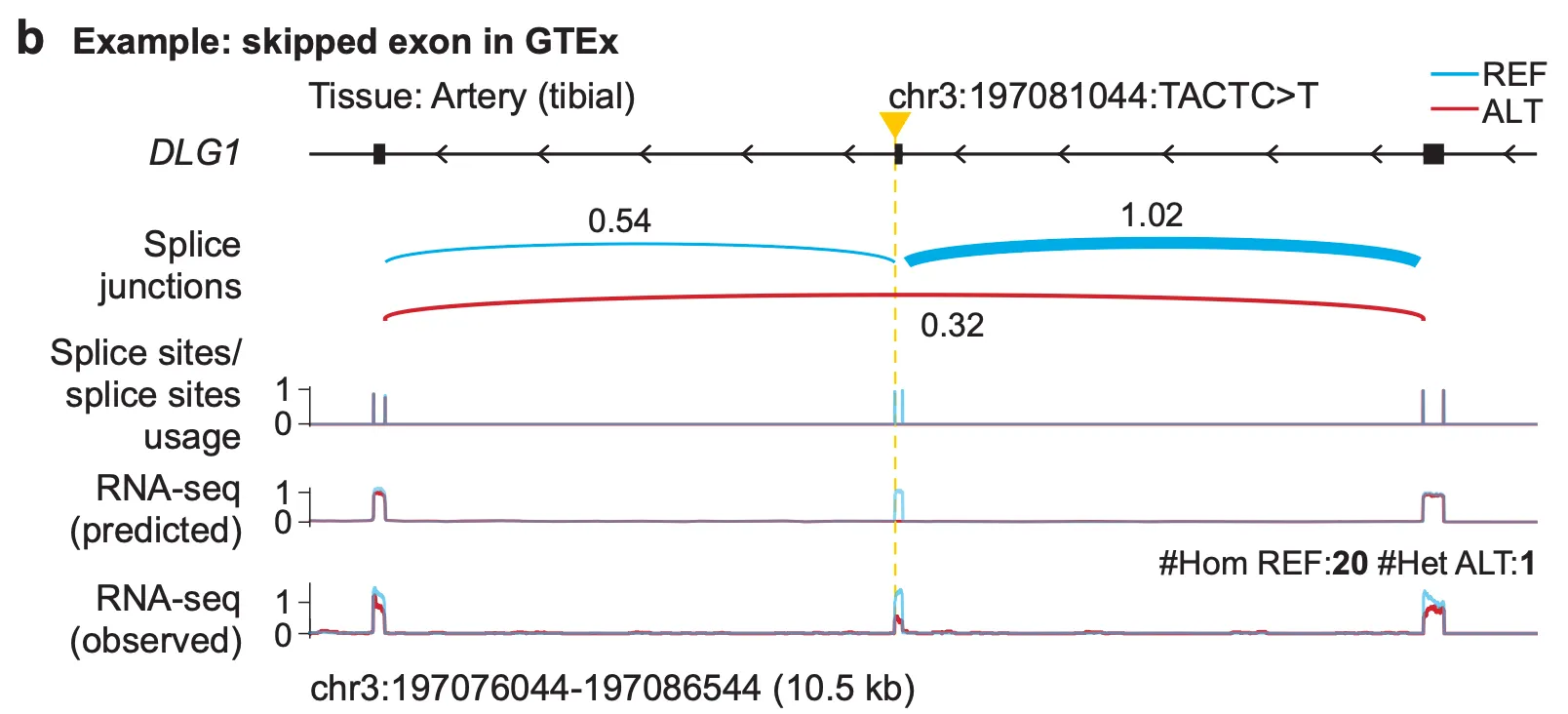
Fig. 3b - 변이에 의한 exon skipping 예측
첫 번째 예시에서는 GTEx 샘플 중 경골동맥(tibial artery) 조직에서 발생하는, chr3:197081044 위치의 4개 염기쌍 결실(TACTC>T)의 효과를 제대로 예측할 수 있는지를 확인해 봅니다. 이 변이는 실제로 근처 exon이 스플라이싱 과정에서 건너뛰어져 mature mRNA에 포함되지 않는 엑손 스키핑(exon skipping)을 유발하는 변이로 알려져 있습니다. AlphaGenome 예측에서 실제로 이 엑손 스키핑을 관찰할 수 있을까요? 결과를 살펴봅시다.
위 그림은 3번 염색체의 197076044번째 염기에서부터 197086544번째 염기까지의 약 10,000bp 정도의 지역(chr3:197076044-197086544)을 확대하여 보여주고 있습니다. 가상의 x축이 있어서 유전체 상의 좌표를 나타낸다고 보시면 됩니다. 그림 안의 파란색 선들은 모두 변이가 발생하지 않은 표준(reference) 상태의 결과이고, 빨간색 선들은 변이가 발생한(alternative) 상태의 결과입니다.
맨 윗줄의 생선가시같은 track은 해당 지역에 존재하는 유전자 DLG1의 annotation을 보여주고 있습니다. 왼쪽으로 향하는 화살표는 이 유전자가 (-) strand 쪽의 서열을 주형으로 갖는 유전자라는 것을 나타내며, 선이 굵은 부분은 exon, 나머지 부분은 intron 영역을 나타냅니다.
두 번째 줄의 “Splice junction” 트랙에서는 스플라이싱이 어떤 패턴으로 이루어지고 있는지를 엿볼 수 있습니다. 곡선과 곡선 위의 숫자는, 곡선의 왼쪽 끝과 오른쪽 끝을 잇는 스플라이싱의 근거가 되는(supporting하는) 시퀀싱 read의 양을 의미합니다(쉽게 말해 해당 read를 참조유전체에 매핑해봤더니 왼쪽 끝과 오른쪽 끝이 저렇게 멀리 떨어져서 매핑된 것이죠). 파란색 곡선은 그림에 나타난 3개의 exon중 1~2번 exon, 2~3번 exon을 순차적으로 방문하고 있는 반면, 빨간색 곡선은 2번 exon을 건너뛰고 1, 3번 exon을 곧바로 연결하고 있습니다. 즉, 2번째 exon의 엑손 스키핑을 의미하고 있는 것이죠.
세 번째 줄의 Splice sites/splice sites usage 트랙도 비슷한 결과를 나타냅니다. 1, 3번 exon의 시작과 끝 부분의 splice site들은 빨간색, 파란색 peak이 모두 올라와있어 해당 splice site가 사용되고 있음을 확인할 수 있는 반면, 2번 exon을 감싸는 splice site들은 표준 상태에서만 사용되고 있음을 알 수 있습니다.
흥미로운 것은 네 번째 줄과 다섯 번째 줄입니다. 이 트랙들은 각각 AlphaGenome이 예측한 RNA-seq track과 실제 관찰된 RNA-seq track을 나타냅니다. (참고로 여기서 실제 관찰된 track은 GTEx 내의 tibial artery 샘플 중 reference allele을 가지는 샘플 20개의 RNA-seq track의 평균과, alternative allele을 가지는 샘플 1개의 RNA-seq 결과를 보여주고 있습니다.) 2번째 exon 영역에 집중해보면, 변이 상태의 AlphaGenome 예측에서는 2번째 exon 영역에 read coverage가 하나도 나타나지 않고 있지만, 표준 상태의 AlphaGenome 예측에서는 적절히 높은 read coverage를 예측하고 있음을 확인할 수 있습니다. 즉, AlphaGenome이 chr3:197081044:TACTC>T 4bp deletion 변이에 의한 exon skipping을 잘 예측하고 있다는 것을 보여주는 예시가 되겠습니다.
예시 2) 변이에 의한 novel splice junction의 도입을 예측
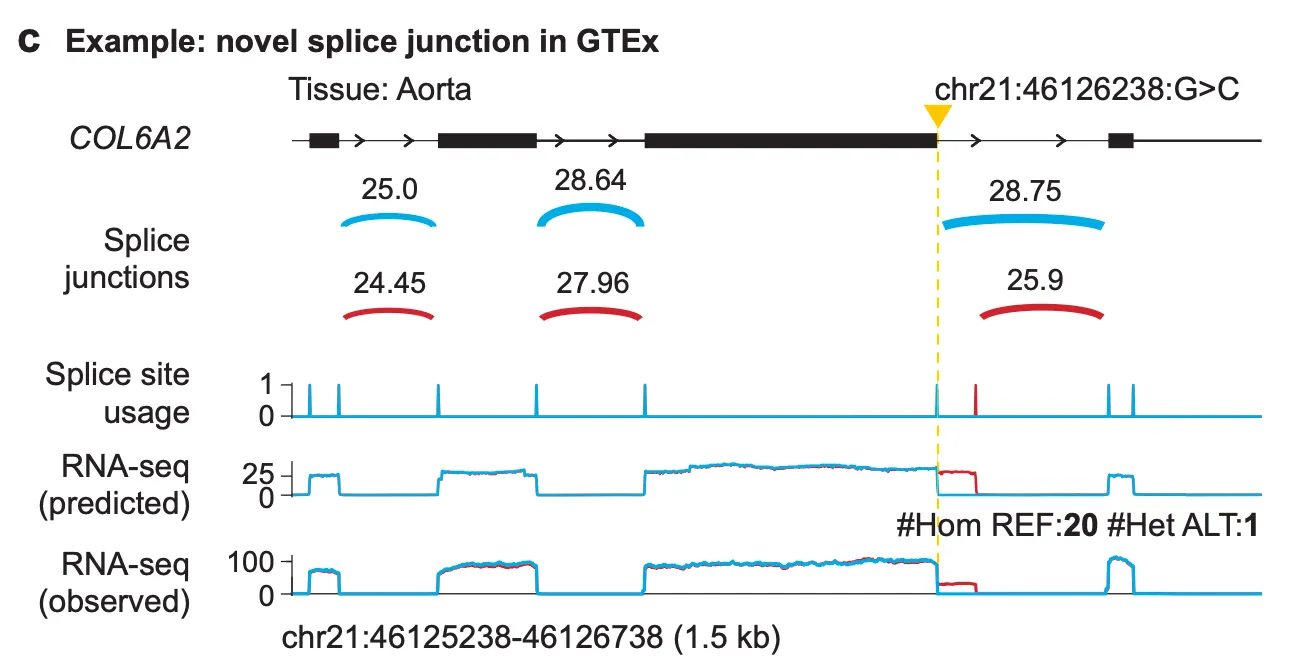
Fig. 3c - 변이에 의한 novel splice junction 예측
이 예시에서는 COL6A2 유전자 영역 내의 chr21:46126238 위치의 G 염기가 C로 바뀌는 변이 (chr21:46126238:G>C)에 의해 새롭게 도입되는 splice junction을 예측할 수 있음을 보여줍니다.
(예시 1)과 같은 방법으로 변이의 효과를 해석해보자면, “변이에 의해 exon 직후의 splice site가 이용되지 못하고, 해당 위치보다 약간 downstream에 존재하는 splice site가 대신 이용되는 상황”으로 이해해볼 수 있겠습니다. 역시 RNA-seq (predicted) 트랙을 보시면, 변이 상태에서만 exon 뒤쪽으로 추가적인 read coverage를 예측하고 있음을 알 수 있고, 이는 곧 novel splice junction이 제대로 예측되었음을 보여주고 있습니다.
예시 3) in silico mutagenesis를 활용한 포괄적인 splice junction 변이 효과 예측
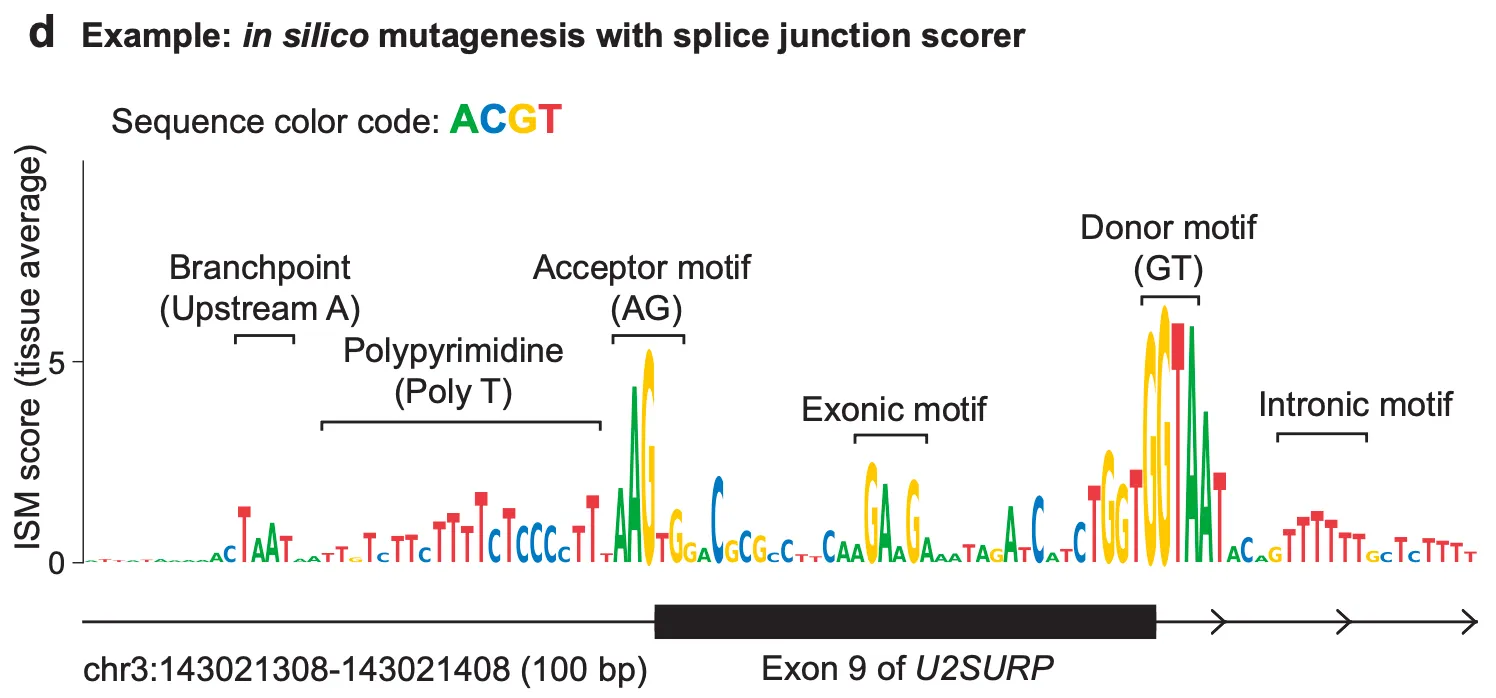
앞선 두 예시에서는 미리 효과가 알려진 변이를 정해 두고, 변이 상태의 AlphaGenome 예측이 우리가 기대하는 효과를 제대로 포착해낼 수 있는지를 확인했다면, 이번 예시에서는 보다 일반적으로 AlphaGenome이 “splicing 관련 motif의 특성”들을 포착해내고 있음을 보여줍니다.
그 방법으로 in silico mutagenesis (ISM)라는 분석 기법을 사용하는데요, 말 그대로 컴퓨터 상에서(in silico) 돌연변이 생성(mutagenesis) 실험을 해보는 것으로 이해하시면 됩니다. 실제 실험실 환경의 saturation mutagenesis 실험과는 달리 AlphaGenome과 같은 AI 모델을 이용하면, 해당 영역 내의 모든 염기를 모든 다른 염기들로 변이시켜 보면서 그 결과를 보는 실험이 비교적 손쉽게 가능하기 때문에, 우리가 관심 있는 output 값 (여기서는 스플라이싱)에 특정 염기가 미치는 영향을 포괄적으로 파악할 수 있게 됩니다.

In silico mutagenesis (ISM)
본 논문 전반에 걸쳐 in silico mutagenesis는 아주 주요한 실험 기법으로 사용되고 있습니다. 위 예시와 같이 각 유전체 위치의 ISM 점수를 나타내는 그림은 다음과 같은 절차로 그려지게 됩니다.
1.
길이 의 입력 서열 내의 각 위치에 대해서 가능한 3개씩의 변이 서열을 모두 생성합니다. (결과적으로 개의 서열이 준비됩니다.)
2.
각 변이 서열을 모두 AlphaGenome 입력으로 넣어 예측 결과를 얻습니다. 해당 예측 결과에서 우리가 관심 있는 유전체 기능 값을 추출해냅니다. (예를 들어, 특정 유전자의 유전자 발현량이라든지, 특정 exon이 스플라이싱에 포함되는 정도라든지..) 그 결과, 각 변이마다 하나의 스칼라 값을 할당할 수 있게 됩니다. 이 값은 “변이의 효과”를 나타내는 값이 되겠습니다.
3.
행렬을 준비합니다. 은 서열 길이를 나타내며, 4개 컬럼은 각각 A, C, G, T 염기를 나타냅니다. 각 서열 위치에서 변이되지 않은 표준 염기 위치에는 0.0 값을 부여하고, 변이된 염기 위치에는 (2) 에서 얻은 변이 효과 스칼라 값을 부여합니다.
4.
개의 행 각각에 대해서, 변이 염기 3개 위치의 값 평균을 구해서 해당 행의 모든 값에서 빼줍니다. 따라서 변이 염기들은 mean-centering되며, 표준 염기 위치에는 0-(변이 염기 값 평균) 값이 채워져 있게 됩니다.
5.
마지막으로, 변이되지 않은 표준 염기 위치에 해당하는 값들을 차례차례 모아서 개의 점수 값을 얻습니다. 이 점수는 우리가 관심 있는 유전체 기능에 대한 해당 표준 염기의 영향력을 의미한다고 볼 수 있겠습니다.
위에서 알아본 ISM 분석 방법 절차를 바탕으로, 위 예시 그림을 해석해보면 ISM score가 큰 염기(높이 솟아 있는 염기)는 곧 “변이되었을 때 U2SURP 유전자의 exon 9의 스플라이싱에 이상이 생기는” 염기라고 볼 수 있고, 실제로 잘 알려진 스플라이싱 모티프들(acceptor, donor, branchpoint 등)의 중요도가 강조되고 있음을 알 수 있습니다.
개별 변이 점수화에 기반한 성능 분석
위 예시들은 AlphaGenome이 잘 작동하는 몇 가지 예시를 보여줄 뿐, 모델의 성능을 객관적으로 판단하기 위한 근거라기에는 무리가 있습니다. 저자들은 AlphaGenome의 예측이 정말로 범용적인 생물학적인 의미를 가지는지 확인하기 위해, 먼저 개별 변이를 점수화하는 방법을 제안합니다.
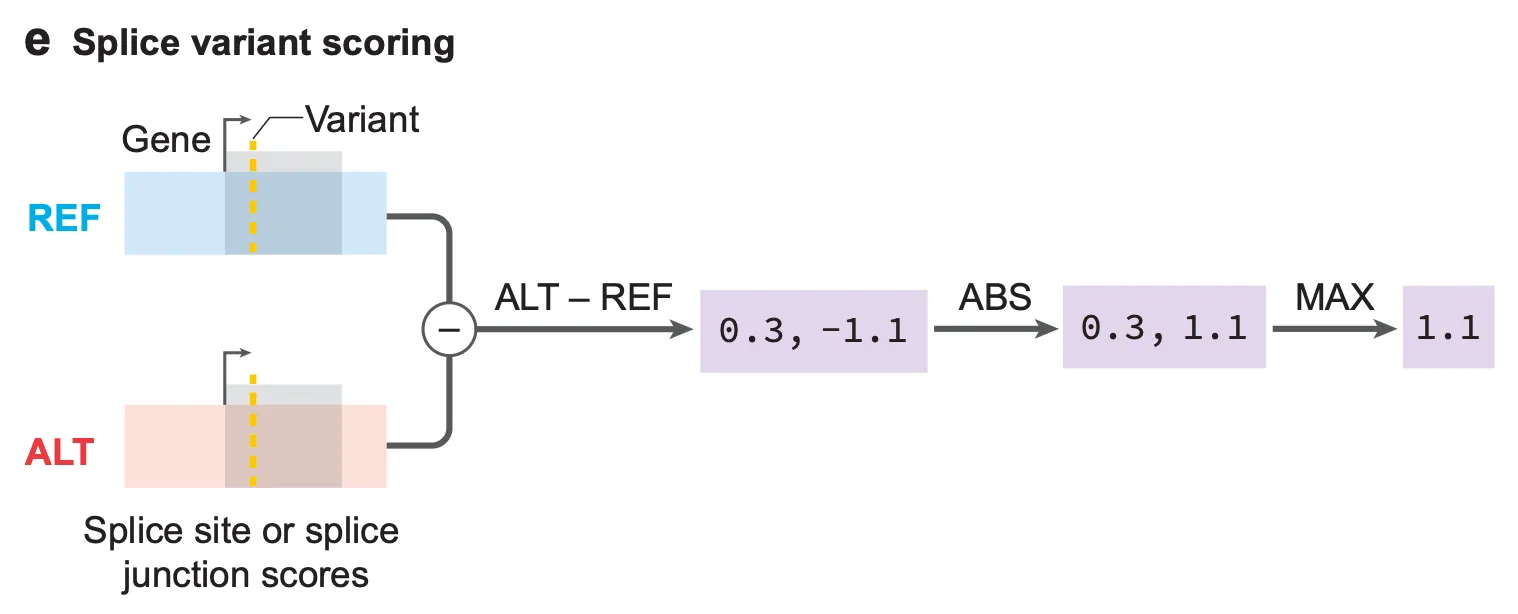
Fig. 3e - 간략하게 도식화된 splice variant scoring 방법.
쉽게 말해, 변이 점수화는 주변 영역(유전자)에 특정 변이가 미치는 가장 큰 영향을 나타내는 점수라고 보시면 됩니다. 변이 점수가 클수록, 주변에 미치는 영향력이 큰 변이가 되겠습니다.
참고) Splice site and splice site usage variant scoring
참고) Splice junction variant scoring
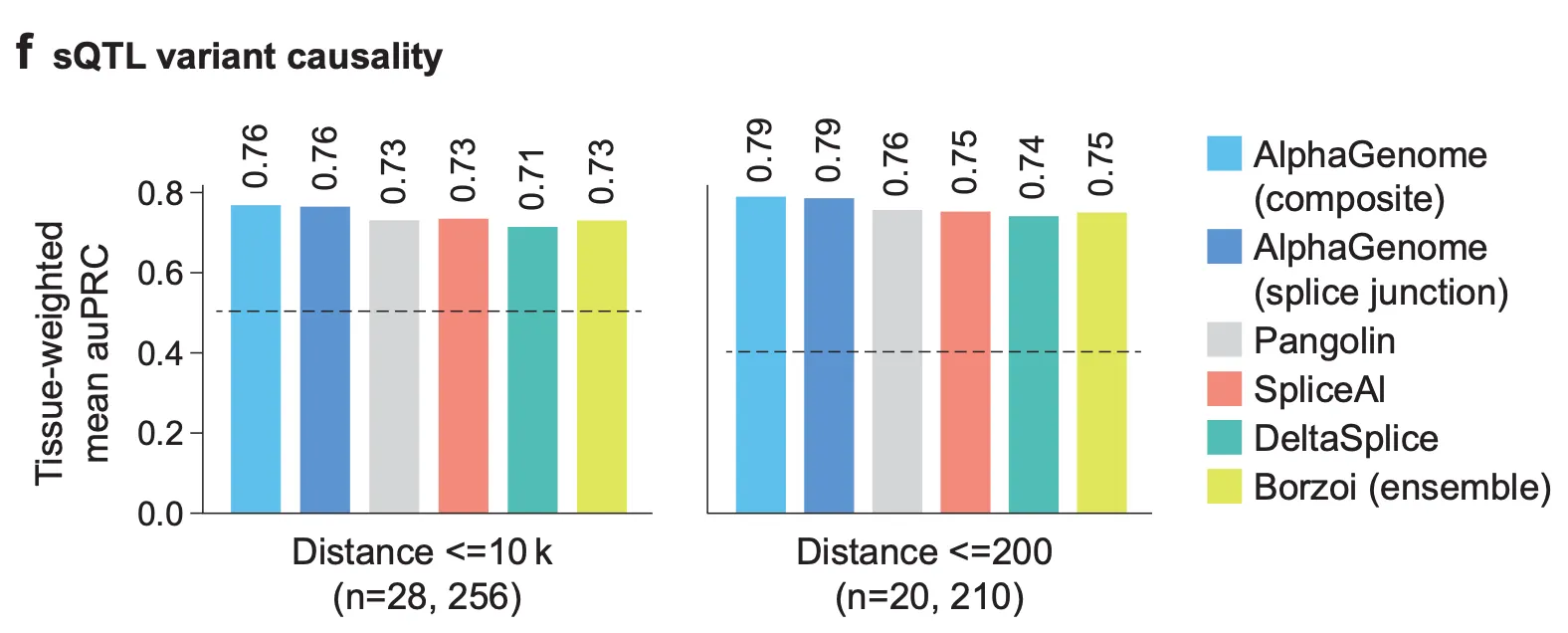
Fig. 3f - sQTL variant causality
현대 유전학의 아버지라 불리는 멘델의 완두콩 실험에 대해 들어보신 적이 있을 겁니다. 완두콩의 색깔, 꼬투리의 모양 등 명확히 구분되는 정성적인(qualitative) 형질이 자손에게 어떻게 전달되는지 연구함으로써 유전의 기본 법칙을 발견한 실험이죠. 완두콩의 경우, 주로 단일 유전자의 DNA 서열 상태(유전형)가 직접적으로 표현형을 결정하는 모델이었기 때문에 잘 통제된 실험이 가능했고, 유전의 기본 법칙을 발견하기에는 아주 안성맞춤인 모델이었다고 할 수 있겠습니다.
하지만 유전학이 분자 수준으로 발전하면서 우리는 특정 유전자의 DNA 서열 상태, 혹은 유전자가 아닌 조절 부위의 DNA 서열 상태가 키, 혈압과 같은 정량적인(quantitative) 형질에 영향을 미친다는 사실을 발견했습니다. 특히 “형질”은 꼭 우리 눈에 보일 필요는 없어서, “특정 유전자의 mRNA 발현량”과 같은 값 또한 형질이라고 볼 수 있습니다. 이렇게 정량적인 형질에 영향을 미치는 유전체상의 위치(loci)를 QTL(quantitative trait loci)라고 정의합니다.
이 결과에서는 “스플라이싱의 정도” 라는 정량적인 형질에 주목하여, 변이가 생겼을 때 특정 유전자의 스플라이싱에 영향을 주는 splicing QTL (sQTL)이 AlphaGenome으로부터 얼마나 정확히 예측되는지를 벤치마크합니다. 주변 유전자에 영향을 주는 이미 알려진 sQTL들은 49개의 GTEx 조직에 대한 eQTL 데이터로부터 curation된 데이터를 이용합니다. 21,514개의 fine-mapped QTL (높은 확률로 causal variant라고 추정된 variant들)과, 같은 수의 distance-matched negative control 데이터로 구성된 데이터라고 하네요.
왼쪽 결과는 스플라이싱이 일어나는 위치와 비교적 멀리 떨어진 (≤10kbp) 위치의 변이를 얼마나 정밀하게 예측하는지를 나타내는 AUPRC 값을 보여주고 있으며, 오른쪽 결과는 스플라이싱 위치와 가까운 변이에 대한 결과입니다. AlphaGenome (composite)은 splice site, splice site usage, 그리고 splice junction에 대한 변이 점수의 합으로 변이의 순위를 매겼을 때의 AUPRC 값을 의미하며, AlphaGenome (splice junction)은 splice junction에 대한 변이 점수만으로 순위를 매겼을 때의 AUPRC 값을 의미합니다. 모든 경우에서 AlphaGenome이 가장 좋은 성능을 보여주고 있네요.
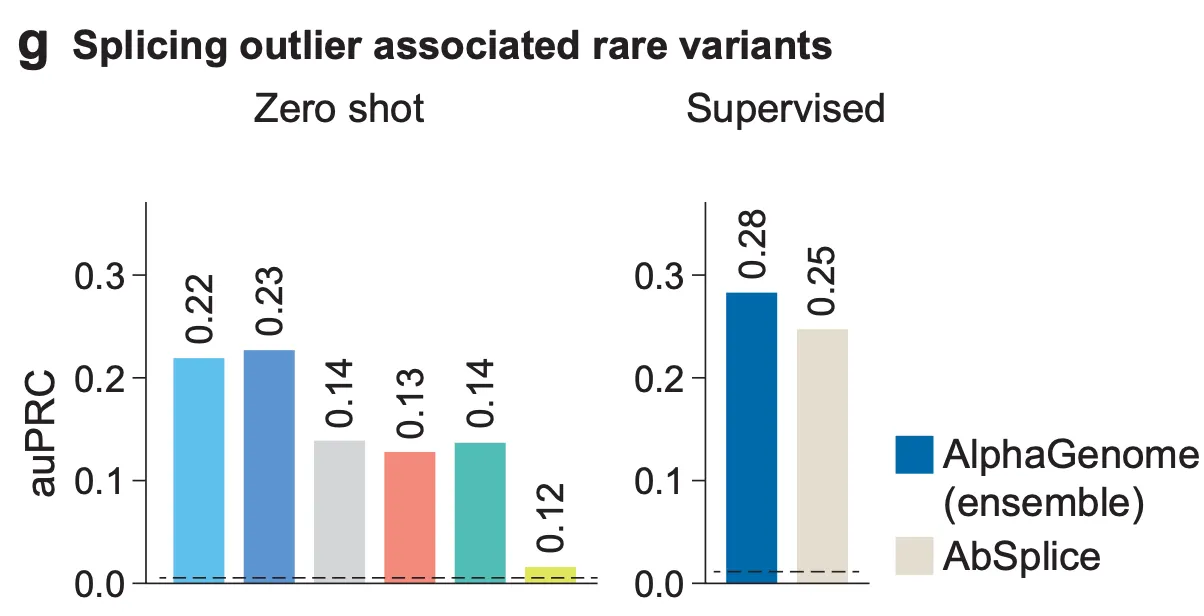
Fig. 3g - 비정상적 스플라이싱과 연관된 희귀 변이 예측.
다음으로, 조금 더 어려운 문제에서는 어떤 성능을 보이는지 알아봅시다. 이 문제는 희귀 변이(rare variant)가 스플라이싱 이상(aberrant splicing)에 연관되어 있는지를 예측하는 문제인데요, 여기서 ‘스플라이싱 이상과 연관된 희귀 변이’란 인구집단에서 빈도 0.1% 미만으로 나타나면서, 적어도 2명 이상에서 나타나는 변이를 의미하며, 동시에 비정상적인 스플라이싱 위치에서 250bp 이하로 떨어져 있는 변이로 정의됩니다. 왼쪽의 “Zero shot” 결과는 AlphaGenome 예측으로부터 직접적으로 얻은 변이 점수 (composite score)로 변이를 예측했을 떄의 결과이며, 오른쪽 결과는 AlphaGenome으로부터 도출된 변이 점수를 포함한 특징(feature)들을 활용하여 Explainable Boosting Classifier 모델을 별도로 학습시켰을 때의 예측 성능입니다. 역시 타 모델에 비하여 스플라이싱과 연관된 희귀 변이에 대한 예측 성능이 높게 나타나는 것을 확인할 수 있습니다.
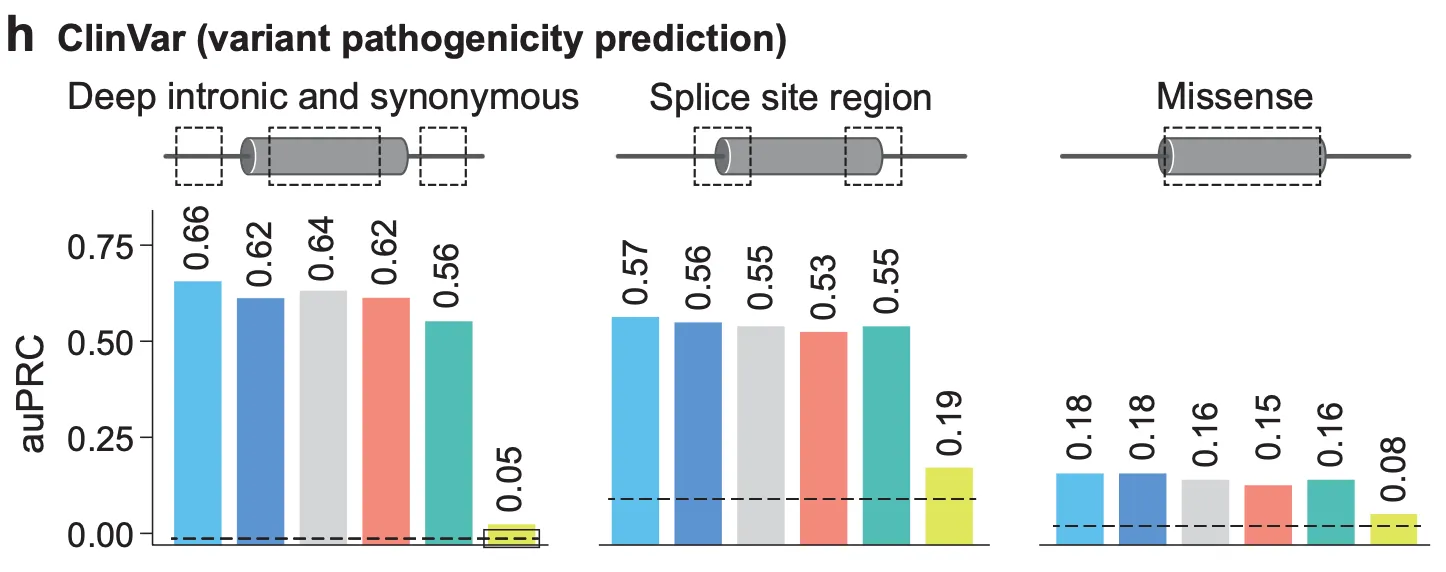
Fig. 3h - ClinVar 변이 병원성 예측.
다음으로, 저자들은 AlphaGenome의 스플라이싱 변이 점수가 실제로 변이의 병원성(pathogenicity)와 얼마나 좋은 연관성을 보이는지를 검증합니다 (Fig. 3h). ClinVar에 존재하는 변이 중 다음 3가지 카테고리에 해당하는 변이들을 수집하여, 해당 변이들의 스플라이싱 변이 점수를 계산하여 순위를 매긴 다음, 병원성 변이(pathogenic variant)와 비병원성 변이(benign variant)를 구분해낼 수 있는 능력을 갖는지를 확인해봅니다.
1.
Deep intronic and synonymous variants: ‘intronic’으로 주석이 달려있으면서 가장 가까운 splice site에서 >6bp 떨어진 변이 혹은 ‘synonymous’으로 주석이 달려 있으면서 가장 가까운 splice site에서 >3bp 떨어진 변이. (1,628 positive / 95,269 negative)
2.
Splice site region: ‘synonymous’, ‘missense’로 주석이 달린 exon 변이 중 splice site에서 <3bp 떨어진 변이 혹은 ‘intronic’으로 주석이 달린 변이 중 splice site에서 <6bp 떨어진 변이. (7,155 positive / 47,354 negative)
3.
Missense variant: ‘missense’로 주석이 달려 있으면서 AlphaMissense가 ‘Likely Benign’으로 예측한 변이(?) (5,108 positive / 100,031 negative)

Clinvar
Clinvar는 유전체 정보의 임상적 해석을 위해 구축된 공개 데이터베이스로, 유전적 변이와 질병 간의 연관성에 대한 해석을 표준화하고 공유하여, 임상유전학 연구 및 진단 분야의 신뢰도를 높이는 것을 목적으로 합니다.
결과적으로, 이 3개 카테고리의 변이에 대해서 모두 경쟁 모델보다 더 좋은 pathogenic variant / benign variant 구분 능력을 갖고 있다는 것을 확인할 수 있습니다.
유전자 발현 변화 예측
eQTL 효과 예측
앞선 스플라이싱 관련 결과에서는 sQTL의 효과를 예측했다면, 유전자 발현과 관련해서는 expression QTL (eQTL)의 효과를 예측합니다. 이름에서 예상하실 수 있듯이, eQTL은 유전자 발현량에 영향을 미치는 유전체상의 위치를 의미합니다. 쉽게 말해, 유전체 상의 위치 X에 존재하는 특정 DNA 염기 하나가 A에서 G로 바뀔 때, 관련된 유전자의 발현량이 유의미하게 증가(혹은 감소)한다면, X는 eQTL이라고 볼 수 있는 겁니다.
얼핏 생각하기에도, 유전체 상의 임의의 염기가 변했을 때 특정 유전자의 발현량이 어떻게 변할지 예측하는 것은 매우 어려운 일입니다. 당연하게도, eQTL의 효과 방향 예측 (eQTL sign prediction; 유전자 발현을 증가시킬 것인지, 감소시킬 것인지), 더 나아가 eQTL의 효과 세기 예측(eQTL effect size prediction)과 같은 문제들은 생물정보학 분야에서 많은 이들이 풀고자 하는 도전적인 문제였습니다.
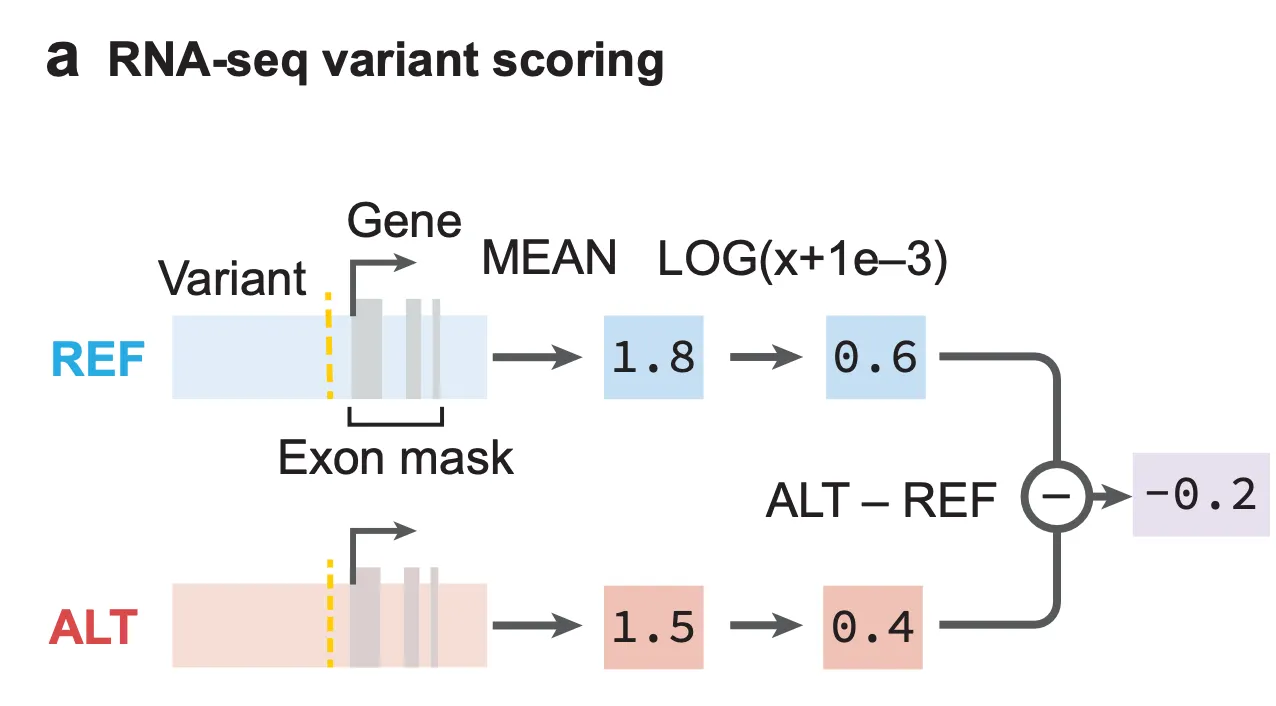
Figure 4a - eQTL 분석을 위한 변이 점수화 방법. Reference allele (REF; 기본형) 상태일 때의 유전자 엑손 부분 RNA-seq track 시그널의 평균 값과, alternative allele (ALT; 변이형) 상태일 때의 시그널 평균 값의 log-fold change로 볼 수 있다.
저자들은 AlphaGenome이 eQTL에 관한 예측들을 잘 해내는지 확인함으로써, 유전자 발현과 연관된 유전체의 문법에 대해 이해하고 있는지를 확인해 봅니다. 가장 먼저 AlphaGenome 기반 eQTL 분석을 위한 변이 점수화 방법을 아래와 같이 정의합니다. 꽤 직관적인 정의 방법인데요, 바로 관심 있는 loci가 reference allele(기본형) 상태일 때의 예측된 유전자 발현량 대비, alternative allele(변이형) 상태일 때의 유전자 발현량이 몇 배 증가/감소했는지를 점수화하는 방식입니다.
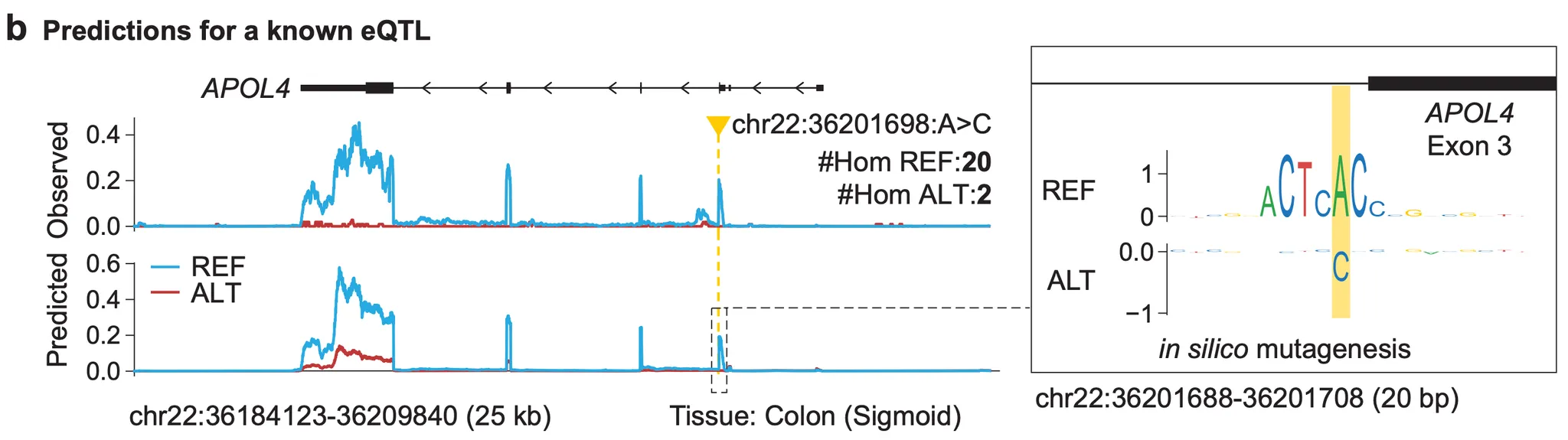
Figure 4b - chr22:36201698:A>C 변이와 연관된 APOL4 유전자 발현량 감소를 예측하는 AlphaGenome.
다음으로, 예시를 하나 보여주네요. eQTL이라 잘 알려진 위치 하나를 AlphaGenome에게 보여주고, 원하는대로 예측이 되는지를 보는 실험입니다. 이 위치(chr22:36201698)는 APOL4라는 유전자의 3번째 exon이 끝나는 부분에 존재하며, 원래 참조유전체에서는 A로 존재하는 위치입니다. S자 결장(sigmoid colon) 조직세포에서 이 위치의 염기가 C로 바뀌면(chr22:36201698:A>C) APOL4 유전자의 발현이 급격하게 낮아지는 것이 잘 알려진 eQTL입니다. 왼쪽의 track 중 아래(predicted)에서, AlphaGenome에 의해 변이형(alternative allele; ALT) 상태에서의 유전자 발현이 기본형(reference allele; REF)상태보다 작게 예측된 것을 확인할 수 있습니다.
오른쪽의 in silico mutagenesis 실험으로 이 현상에 대한 해석 또한 보여줍니다. 앞서 splicing 섹션에서 확인했듯이, exon이 끝나는 3’ 말단에는 GT 서열이 존재하여 splice donor 모티프로써 기능합니다. 마침 우리의 eQTL이 “GT” 서열의 “T”에 해당하는 위치였네요. (APOL4 유전자는 유전체의 (-) strand에 존재하여, 그림의 오른쪽이 5’, 왼쪽이 3’입니다.) 여기서의 in silico mutagenesis 결과는 이렇게 해석하면 됩니다: “다른 염기로 변했을 때 유전자 발현량이 많이 감소하는 염기일수록 크기가 위로 크고, 많이 증가하는 염기일수록 아래로 크다”. 이를 바탕으로 위 결과를 해석해보면, 기본형 상태(A)에서는 해당 eQTL 위치를 포함하는 splice donor의(AC → reverse complement 하여 생각해보면 GT입니다) 중요도가 매우 크게 잡혔지만, ALT 상태에서는 그렇지 않음을 알 수 있습니다. 즉, 예측된 발현량 감소의 잠재적인 메커니즘을 잘못된 splicing 때문이라고 짐작해볼 수 있는 것이겠죠!
개인적인 궁금증으로는, 결국 5’-GT-3’ splice donor motif를 5’-GG-3’ 으로 바꾸는 것이니 aberrant splicing이 일어나는 것 까지는 이해가 되는데, 그러면 intron 부분의 expression이 올라간다거나 하는 결과가 나와야 하지 않을까 싶기는 합니다. 왜 전체적인 유전자 발현량이 감소한다고 예측이 되는 걸까요?
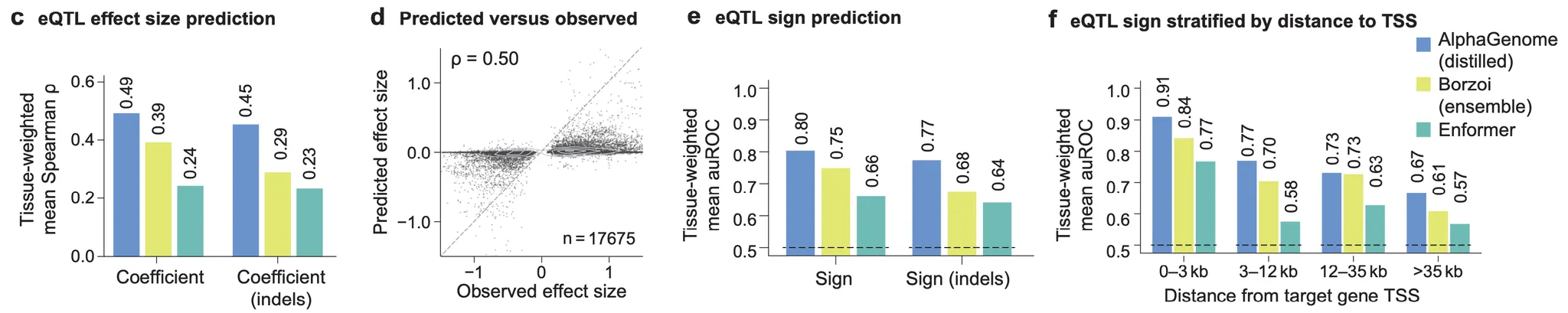
Figure 4c-f - eQTL 효과 관련 예측 성능
다음으로는, 위와 같은 eQTL 효과 예측의 일반적인 성능은 어떤지 측정해 봅니다. 먼저 가장 왼쪽, 그림 4c는 AlphaGenome이 예측한 eQTL 점수(위의 변이 점수화 방법을 참고해주세요)와 eQTL effect size 간의 상관계수를 나타냅니다. Borzoi, Enformer보다 월등한 성능을 보여주고 있네요. 그림 4d는 해당 상관계수를 얻었을 때의 산포도를 보여줍니다. 얼핏 보기에는 생각보다 “예쁜” 상관관계는 아니고, 또한 effect size를 0으로 예측하여 틀리는 경우가 굉장히 많다는 것을 알 수가 있네요.
위 분석에서 일종의 “정답” 값으로 사용한 eQTL effect size(SuSiE beta posterior)는 어떤 값이며, 어떻게 구했는지 궁금하신 분들은 아래를 참고해주세요.

SuSiE posterior inclusion probability 및 beta posterior 값의 해석
그림 4e와 4f의 경우는 해당 eQTL이 유전자 발현을 증가시키느냐, 감소시키느냐의 방향을 예측하는 성능이 AlphaGenome에서 가장 좋음을 보여주고 있습니다. 왼쪽의 그림 4e는 전체 eQTL에 대한 성능이며, 오른쪽의 4f는 유전자의 전사 시작부위(transcription start site; TSS)에서부터 eQTL까지의 거리를 기준으로 성능을 나누어 확인해본 결과입니다. 확실히 유전자와 eQTL 사이의 거리가 멀수록 eQTL 효과 방향 예측이 어려워짐을 알 수가 있네요.
참고로, 그림 상에 (indels)로 표시된 부분은, 1개의 기본형(REF) 상태 염기가 변이형(ALT) 상태가 되면서 2개 이상의 염기가 되거나(삽입; insertion), 제거되는 경우(결실; deletion)에 한정한 성능을 나타냅니다.
인핸서 - 유전자 연결(enhancer-gene linking) 예측 성능
어떤 유전자의 발현은 유전체 상에서 해당 유전자에 가까운 조절 요소들(예: 프로모터 부위의 히스톤 변형 및 염색질 접근성)의 영향을 가장 많이 받습니다. 그러나 신기하게도, 많은 경우에 유전자와 멀리 떨어진 부위에 의해서도 유전자 발현이 조절되는 현상이 알려져 있습니다. 오른쪽 그림에서 볼 수 있듯이, 이들은 1차원적으로는 유전자와 멀리 떨어져 있지만 염색질 접힘(chromatin folding)에 의해 유전자와 가깝게 위치하고, 전사 인자들을 추가적으로 끌여들여 유전자 발현을 조절합니다.
이러한 유전체 부위들을 통틀어 원거리 조절 요소(distal control elements/distal regulatory elements)라 부르며, 그 중에서도 유전자 발현을 촉진하는 요소들을 인핸서(enhancer), 억제하는 요소들을 사일런서(silencer)라 합니다. 아무래도 역사적으로 유전자 발현을 촉진하는 인핸서의 중요성이 부각되어 온 만큼, 축적된 데이터와 연구 결과 또한 인핸서 쪽이 더 많은 것이 사실입니다. 여기서는 인핸서에 주목해보도록 하겠습니다.
인핸서가 특정 조직에서 정해진 유전자의 발현을 정교하게 조절하는 것은 조직 특이적인 유전자 발현에 중요합니다. 반대로, 특정 조직에서 어떤 유전자와 연결되지 말아야 할 인핸서가 잘못 연결되어 해당 유전자 발현을 촉진할 경우 질병의 원인이 되기도 합니다. 결국 어떤 조직에서 특정 유전자와 연결된 인핸서를 찾아내고, 나아가 인핸서-유전자 연결을 예측하는 것은 질병의 기작을 이해하는데 굉장히 중요하다고 할 수 있겠습니다.
인핸서에 의한 유전자 발현 조절. 출처: https://opened.cuny.edu/courseware/lesson/685/student-old/?task=3
저자들은 AlphaGenome이 서열로부터 인핸서-프로모터 상호작용(E-P interaction)을 성공적으로 예측할 수 있다는 벤치마크 결과를 보여줍니다. 벤치마크 데이터로는 ENCODE-rE2G 논문에서 잘 정리해서 사용한, CRISPRi 실험을 통해 검증된 인핸서-유전자 쌍을 사용합니다. 총 10353개의 쌍들 중 471개의 정답 쌍이 있는 데이터라고 하네요. CRISPR를 사용한 enhancer 스크리닝 기법(예를 들어, CRISPRi-FlowFISH)은 흔히 사용되는 기법이니, 궁금하신 분들은 아래의 설명을 참고해 주세요.

Enhancer-gene pair 검출을 위한 CRISPR 실험

CRISPRi-FlowFISH 실험 기법

ENCODE-rE2G 벤치마크 데이터셋
그러면 AlphaGenome을 통해서 어떻게 인핸서-프로모터 상호작용을 예측할 수 있을까요? 여기서는 gradient 기반 방법을 사용했다고 합니다. 방법을 간단히 정리하면 다음과 같습니다.
1.
타겟 유전자가 중심으로 오게 윈도우를 위치시킨 다음, 해당 서열을 AlphaGenome에 넣어 유전자 발현량(RNA-seq) track을 예측합니다. (K562 cell line track에 대해서 수행합니다.)
2.
엑손 부분의 유전자 발현량 값의 평균을 구한 다음, 이 값으로 backpropagation을 수행하여 입력 서열에 걸리는 gradient contribution score를 구합니다.
•
즉, 각 입력 서열 위치에 대해 “염기를 다른 염기로 대체했을 때 유전자 발현량이 얼마나 변하는지”를 나타내는 값이라고 보면 됩니다.
3.
관심 있는 후보 인핸서를 중심으로 하여 2400kbp 윈도우를 잡고, gradient contribution score를 구합니다. 중심 부분의 점수를 더 강조하기 위해, 표준편차 300의 정규분포 가중치를 줍니다.
4.
이렇게 해당 유전자에 대해, 주변에 존재하는 후보 인핸서들의 점수를 뽑아낼 수 있습니다. 점수가 높을수록 유전자와 연관성이 높은 인핸서로 예측했다는 의미입니다.
이제 결과 그림을 한 번 살펴봅시다 (아래 그림). 먼저 zero-shot 성능이 비교 대상 모델인 Borzoi 보다 약간 높아졌음을 보여주고 있네요 (아래 그림, 왼쪽). 여기서 zero-shot이라 함은, 명시적으로 “이 인핸서와 이 유전자가 쌍을 이룬다”라는 정보를 주고 지도학습을 한 것이 아니라, 유전자 발현 및 여타 track들을 예측하는 (관련성은 있지만 다른) 태스크로 학습한 모델을 인핸서-유전자 쌍 예측에 활용했다는 의미입니다. Y축(area under precision-recall curve)은 아주아주 쉽게 말해, 총 10353개의 쌍들 중 471개의 정답 쌍들의 점수가 얼마나 높게 분포하는지에 대한 값으로 보시면 됩니다. 예측 성능이죠. 100kb보다 멀리 떨어진 인핸서에 대한 예측은 아직 만족스러운 수준은 아닌 것으로 보이네요.
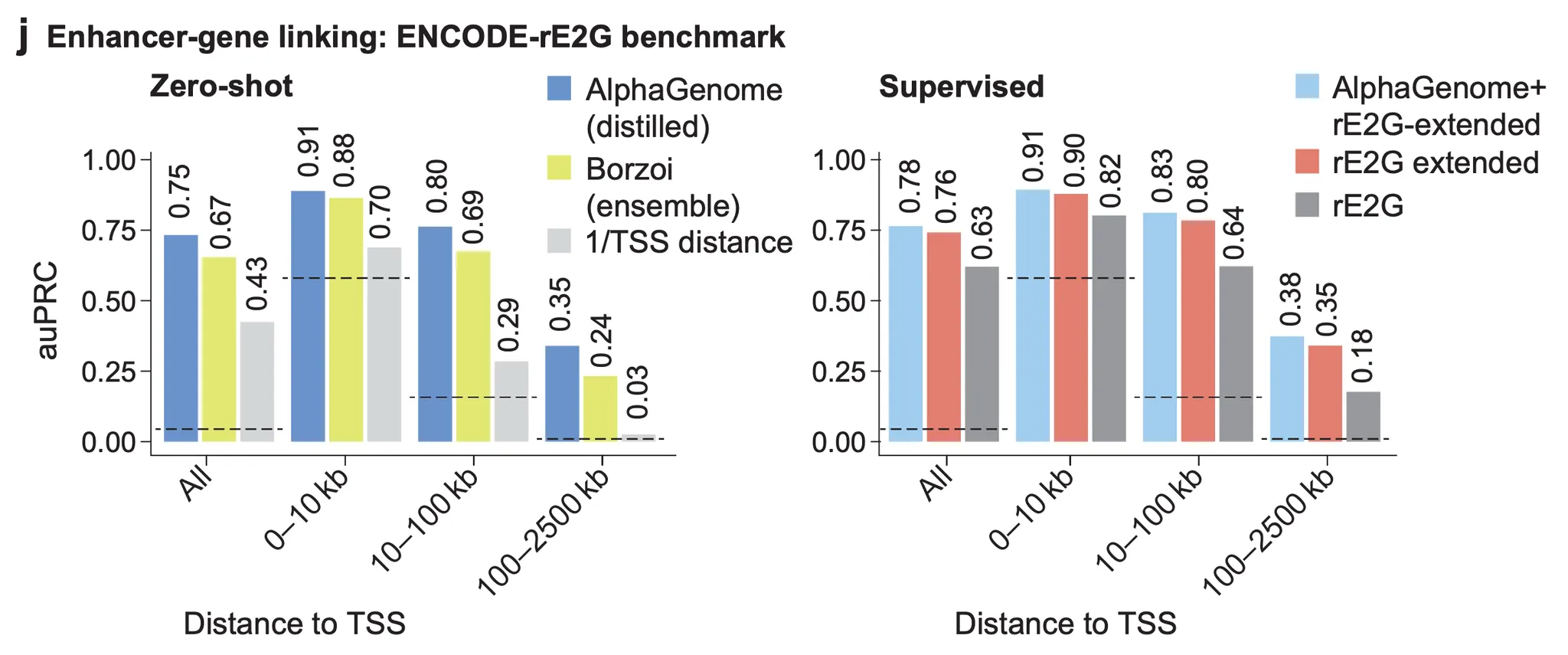
Figure 4j - Zero-shot 예측 성능 (왼쪽), ENCODE-rE2G-extended 모델의 입력 피처에 AlphaGenome에서 얻은 인핸서-유전자 예측 점수를 함께 사용했을 때의 예측 성능 (오른쪽)
오른쪽 그래프는 ENCODE-rE2G-extended 라는 인핸서-유전자 쌍 예측 모델이 학습할 때 사용했던 입력 피처와, AlphaGenome에서 얻은 인핸서-유전자 예측 점수를 함께 사용했을 때 예측 성능이 향상됨을 보여주고 있습니다. ENCODE-rE2G-extended 모델이 원래 사용했던 입력 피처는 염색질 접근성, 3차원 염색질 접촉 빈도 등 직관적으로 이해가 가능한 handcrafted 피처들입니다. 여기에 AlphaGenome이 학습을 통해 얻은 피처를 더했을 때 추가적인 성능 향상이 있다는 것은, AlphaGenome의 멀티모달 학습을 통해 형성된 유전자 발현의 원거리 조절에 관한 내재적인 피처가 기존의 handcrafted 피처들이 포착하지 못한 무언가를 잡아내고 있다는 것으로 생각해볼 수 있겠습니다.
끝으로, Zero-shot AlphaGenome 모델의 성능이 supervised AlphaGenome + rE2G-extended 성능에 많이 뒤떨어지지 않는다는 점 또한 강조하고 있습니다. 왼쪽 그래프와 오른쪽 그래프의 수치들을 비교해보면 되겠네요.
변이에 의한 대안적 폴리아데닐화(alternative polyadenylation) 양상 변화 예측
성숙한 mRNA(mature mRNA)의 말단에는 A염기가 반복되어 이어진 폴리-A 꼬리(poly-A tail)이 존재합니다. 이 꼬리는 전사 과정에서 유전자에 존재하는 폴리아데닐화 신호(polyadenylation signal)서열을 인식하여 일련의 단백질들이 결합하고, 그로부터 10~35bp 정도 지난 부분에서 절단(cleavage)이 일어난 직후 폴리-A 중합효소(Poly(A) polymerase)가 폴리-A 꼬리를 합성함으로써 형성됩니다.
한 유전자에는 여러 개의 폴리아데닐화 신호가 존재합니다. 어떤 신호를 인식하느냐에 따라 폴리-A 꼬리가 시작되는 지점이 달라지며, 결과적으로 mRNA의 3’ UTR 서열이 달라지게 됩니다. 3’ UTR 서열은 mRNA의 조절에 관여하는 여러 서열들이 존재하기 때문에(예를 들어, miRNA가 결합하는 부위라던지), 이러한 대안적 폴리아데닐화(alternative polyadenylation; APA)는 mRNA 반감기 조절 혹은 조직 특이성(tissue specificity)와 깊은 연관을 맺고 있습니다.
AlphaGenome의 RNA-seq track 예측은 이러한 대안적 폴리아데닐화까지 모두 반영된 결과일까요? 저자들은 이 질문에 답하기 위해 폴리아데닐화 변이 점수(polyadenylation variant score)를 고안하여 polyadenylation QTL (paQTL)들을 예측할 수 있는지 평가합니다.

AlphaGenome을 활용한 폴리아데닐화 변이 점수 계산
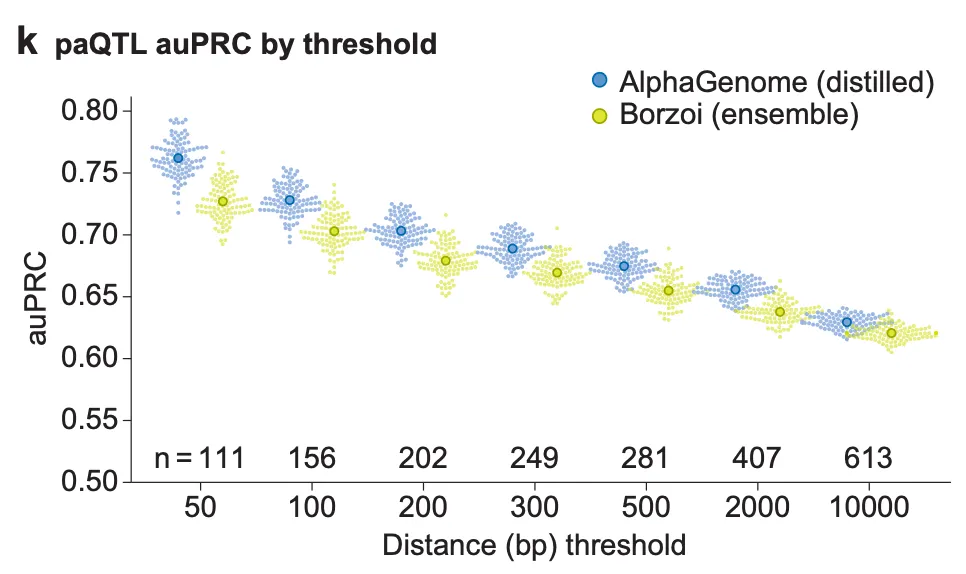
위 그림은 polyadenylation site로부터의 거리에 따른 paQTL 예측 성능을 보여줍니다. Polyadenylation site로부터의 거리가 멀어질수록 (distal paQTL) 예측 성능이 하락하는 것을 확인할 수 있지만, 모든 거리 그룹에서 AlphaGenome이 Borzoi보다 좋은 성능을 보이고 있군요.
변이에 의한 염색질 접근성(chromatin accessibility) 및 전사 인자 결합(transcription factor binding) 변화 예측
다음으로, AlphaGenome이 과연 특정 변이가 염색질 접근성과 전사 인자 결합에 미치는 영향을 예측할 수 있는지 확인해 봅니다. 저자들은 염색질 접근성에 영향을 미치는 유전체 위치들을 분석하기 위해 chromatin accessibility QTL (caQTL), DNase sensitivity QTL (dsQTL), 그리고 binding QTL (bQTL)에 주목하고 있는데요, 역시 AlphaGenome 예측을 활용하여 변이를 점수화하는 방법부터 정의해 봐야겠죠.
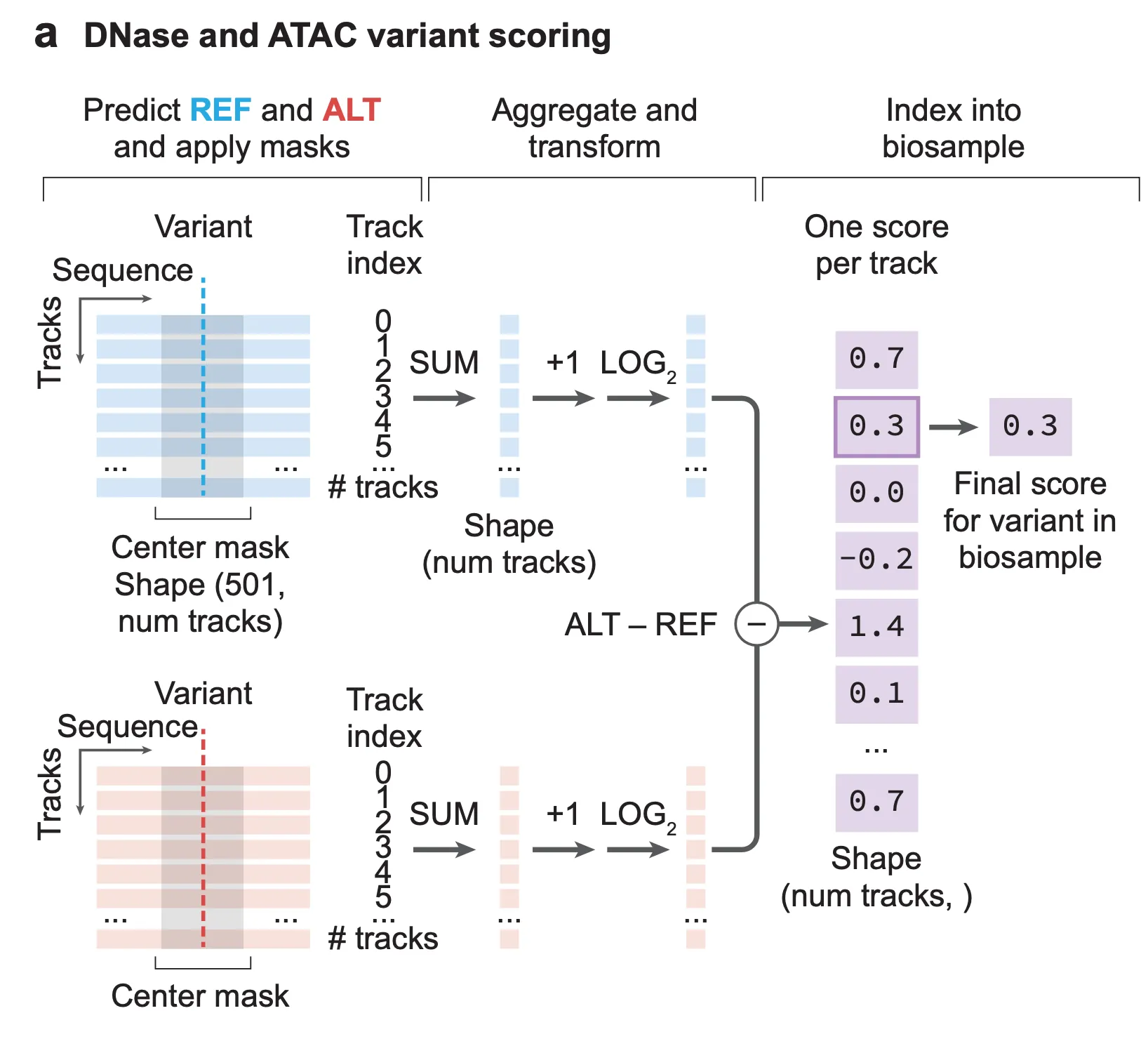
Supplementary Figure 12a. 염색질 접근성(chromatin accessibility)에 variant가 미치는 영향을 점수화하는 방법.
위 그림은 염색질 접근성에 특정 변이가 미치는 영향을 점수화하는 방법을 나타냅니다. 변이를 중심으로 AlphaGenome 예측을 수행한 이후, DNase 및 ATAC track에 대해서 변이를 중심으로 하는 501bp window를 생각합니다. 이 window 내의 시그널 값들을 모두 합한 이후, log2 fold-change를 구하여 이를 점수로 사용하게 됩니다.
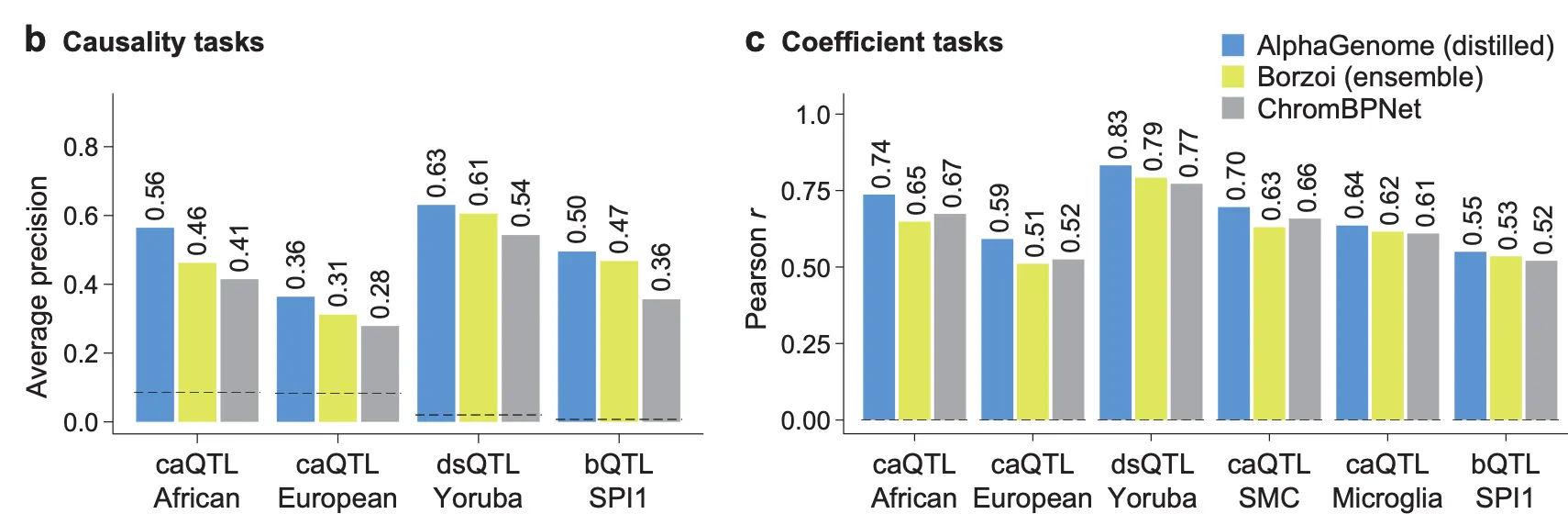
Fig. 5b and c - Chromatin accessibility 및 TF binding QTL 예측 성능
위 그림은 다양한 혈통에서 밝혀진 caQTL, dsQTL 들을 AlphaGenome 변이 점수로 잘 예측해낼 수 있는지를 보여줍니다. 왼쪽의 그림에서는 특정 변이가 과연 실제로 chromatin accessibility 변화를 일으키는지, 인과 관계(causaility)를 잘 예측하고 있는지를 보여주며, 오른쪽 그림에서는 특정 변이가 얼마나 많이 chromatin accessibility의 변화를 가져오는지를 변이 점수로 예측할 수 있는지, 그 상관관계를 보여주고 있습니다. Baseline으로 사용된 ChromBPNet의 경우 염색질 접근성 프로파일을 예측하는 데에 특화된 모델임에도 불구하고 AlphaGenome이 더 뛰어난 성능을 보여준다는 점이 눈에 띄네요.
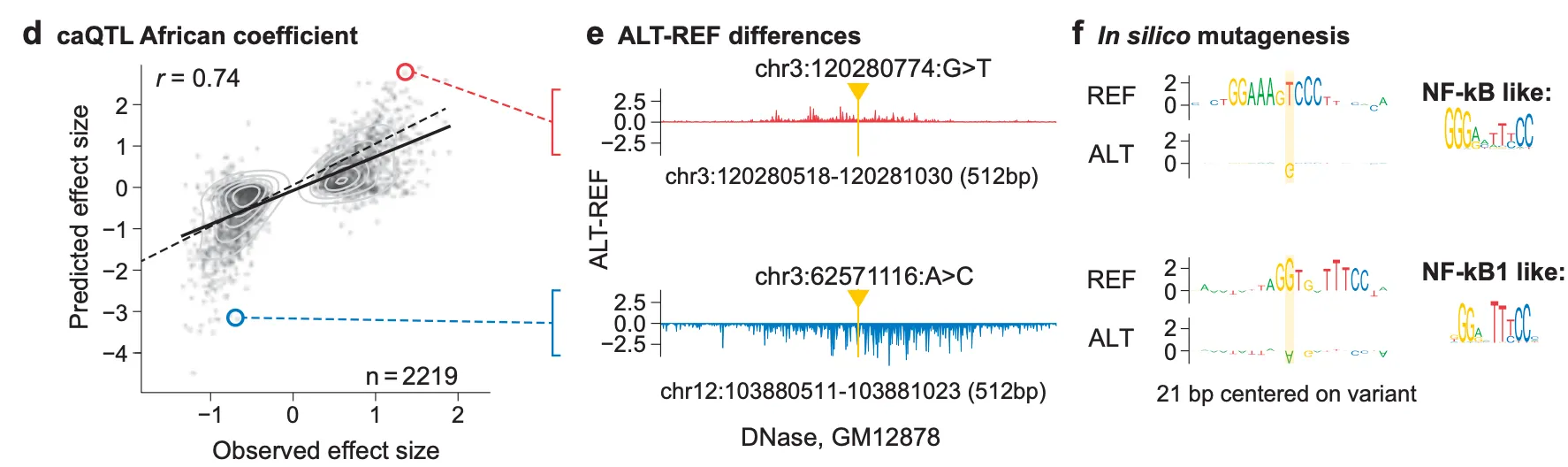
Fig. 5d-f - Case study: 큰 effect size를 가질 것으로 예측되었고, 실제로 그런 caQTL variant 2개 예시.
Fig. 5d는 아프리카계 혈통에서 밝혀진 caQTL들에 대해서, AlphaGenome이 예측한 effect size와 실제 관찰된 effect size 사이에 매우 높은 상관관계가 있음을 보여주고 있습니다. Fig. 5e와 f는 그 중 effect size가 큰 예시 2개를 가지고 case study를 한 결과입니다.
빨간색 예시(chr3:120280774:G>T)는 effect size가 양수인 점으로 미루어볼 때 염색질 접근성을 증가시키는 변이입니다. Fig. 5e (위) 의 ALT-REF 값이 양수라는 점은 실제로 ALT (변이 상태)에서 예측된 DNase-seq track의 coverage가 더 크다는 것을 나타냅니다. Fig. 5f (위)의 ISM 결과에서 REF 상태에서 해당 위치(실제로는 G이지만)가 T로 바뀌었을 때 chromatin accessibility가 더 커지는 것으로 예측됨을 다시 확인할 수 있고, 이렇게 T로 변이되었을 때 해당 위치 근처에 NF-kB 전사 인자가 선호하는 염기서열 모티프가 형성되는 점을 강조하고 있습니다. 실제로 NF-kB는 히스톤 아세틸전이효소(histone acetyltransferase; HAT)나 SWI/SNF 복합체 등을 근처로 불러들여 해당 위치의 염색질 접근성을 높이는 전사 인자로 알려져 있습니다. 즉, 저자들은 해당 변이로 인해 NF-kB가 결합하고, 이 결합으로 인해 변화하는 염색질 접근성까지 예측할 수 있음을 주장하고 있는 것입니다. 파란색 예시(chr3:62571116:A>C라고 되어 있지만.. 아마 오타인 듯 합니다..!)는 반대로 NF-kB1 이 선호하는 모티프를 망가뜨려 결과적으로 염색질 접근성이 낮아진다는 기작을 제시하고 있네요.

Fig. 5g-i - Case study: 큰 effect size를 가질 것으로 예측되었고, 실제로 그런 bQTL variant 2개 예시.
Fig. 5g도 유사하게 이해해볼 수 있습니다. 여기서는 SPI1 전사 인자의 결합에 주목하여, 해당 전사 인자의 결합을 촉진하거나, 억제하는 bQTL들의 예시를 보여주고 있습니다. 빨간색 예시(chr3:62571116:A>C)는 해당 변이에 의해 SPI1 TF-ChIP-seq track의 coverage가 더 높게 예측됨을 나타내며, Fig. 5i에서 실제로 이 변이에 의해 SPI1/SPI-B가 선호하는 염기서열 모티프가 형성됨을 보여줍니다. 파란색 예시(chr12:26131017:A>G)는 반대로 SPI1 모티프가 망가져 SPI1 TF-ChIP-seq track의 coverage가 낮아지는 것을 보여주는 예시가 되겠습니다.
Variant effect의 멀티모달(multi-modal) 해석
저자들은 다음으로 AlphaGenome을 통해서 특정 variant의 효과를 멀티모달(multi-modal)하게 해석할 수 있는지 확인하였습니다. 이를 위한 case study로서, TAL1 유전자의 과발현을 유발하는 세 종류의 변이 집합을 예시로 들었습니다.

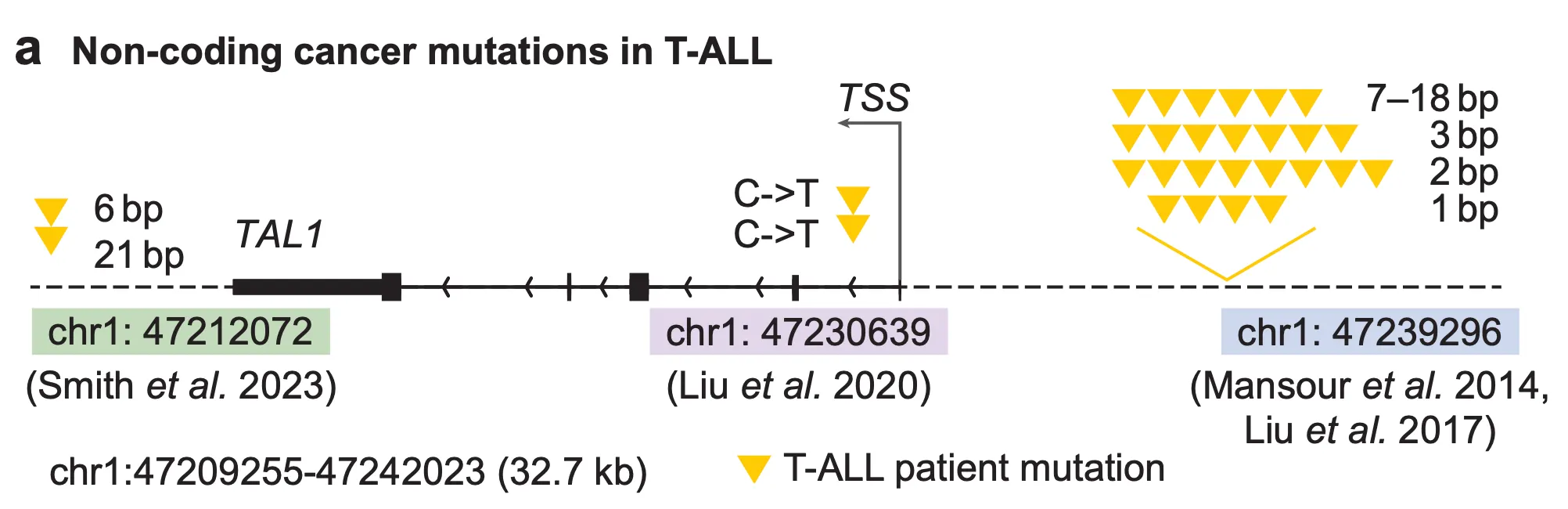
Fig. 6a - T-ALL에서 호발하는 비암호화(non-coding) 변이 목록.
첫 번째 변이 집합은 위 그림 가장 오른쪽에 있는 TAL1 전사 시작 부위(transcription start site, TSS) 앞에 있는 네오-인핸서(neo-enhancer) 변이들입니다. 단일 염기 변이들도 있지만, 2bp, 3bp, 7-18bp등 짧은 DNA 조각들이 삽입(insertion)된 경우도 있군요. 다음으로는 그 왼편의 인트론(intron)에 존재하는 단일염기 변이가 있고, 마지막으로 가장 왼편의 TAL1 유전자 뒤에 존재하는 네오-인핸서 변이들이 있네요. 이러한 변이들은 공통적으로 TAL1 유전자의 과발현을 유도한다고 알려져 있습니다. 과연 AlphaGenome은 이 변이들에 의해 TAL1 유전자의 발현량이 증가한다는 것을 예측할 수 있었을까요?
실험을 위해 저자들은 AlphaGenome으로 예측 가능한 track들 중 T-ALL 세포와 가장 가까운 조상인 “CD34+ common myeloid progenitor” track에 주목했습니다. 가장 먼저 TAL1 앞의 네오-인핸서 유발 변이 중 하나인 chr1:47239296C>ACG 변이를 AlphaGenome에게 보여주자 아래 그림과 같은 흥미로운 해석을 보여주었습니다.
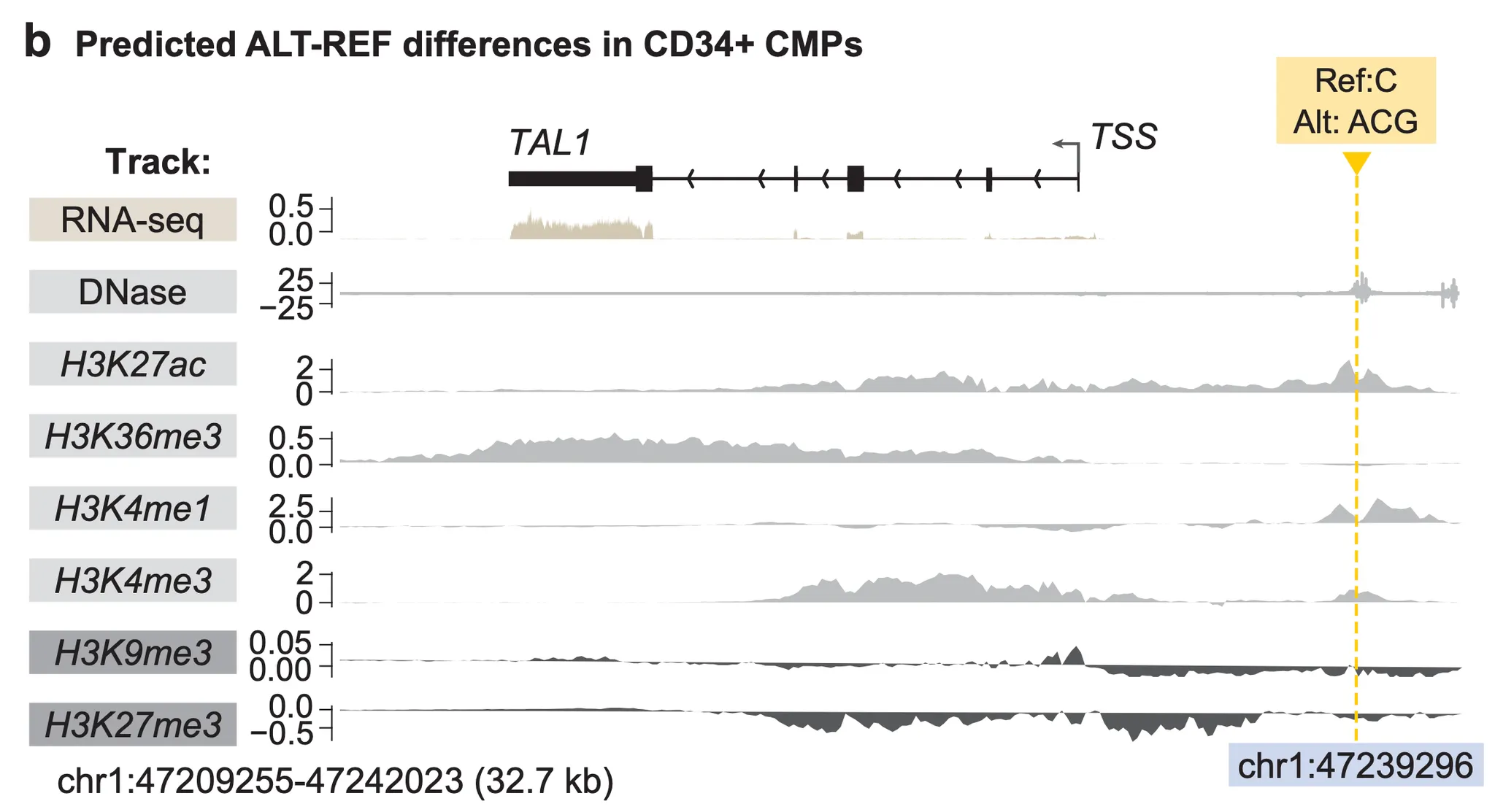
Fig. 6b - chr1:47239296C>ACG 변이에 의한 효과 예측. Track이 위로 올라올수록 변이에 의해 시그널이 더 강해진다는 의미이다.
위 그림에서, 가장 먼저 눈에 띄는 것은 변이에 의해서 TAL1 유전자에 잡히는 RNA-seq 시그널(맨 위)이 더 커졌다는 겁니다. 즉, 유전자 발현이 더 많이 될 것으로 예측한다는 것이죠. 그렇다면 ‘유전자 발현’이라는 결과에 더해, “왜 발현이 증가했을까?”에 대한, 보다 근본적인 해석도 찾아볼 수 있을까요?
놀랍게도, 나머지 히스톤 마크(histone mark)에 대한 track들이 이를 잘 설명합니다! 유전자 발현 활성화와 연관되어 있다고 잘 알려진 히스톤 마크 3종(H3K27ac, H3K4me1, H3K4me3) track을 잘 살펴보면, 변이 근처에서 시그널이 강해진 것을 확인할 수 있습니다. 특히 H3K4me1 및 H3K27ac는 인핸서를 나타내는 히스톤 마크이기 때문에, 해당 지역이 변이에 의한 네오-인핸서로서 기능하고 있다는 것을 알 수 있습니다. 유전자 억제와 연관된 히스톤 마크(H3K9me3, H3K27me3) 시그널들이 변이 근처에서 약해진 점, 그리고 유전자 발현 시 유전자 본체(gene body)지역에서 시그널이 강해지는 H3K36me3 시그널이 강해진 점 또한 해당 지역이 네오-인핸서로써 작동하고 있다는 점을 뒷받침합니다.
나머지 변이에 대해서도 비슷한 결과가 나왔을지 궁금한데요, 아래 그림을 보면 그 결과를 알 수 있습니다.
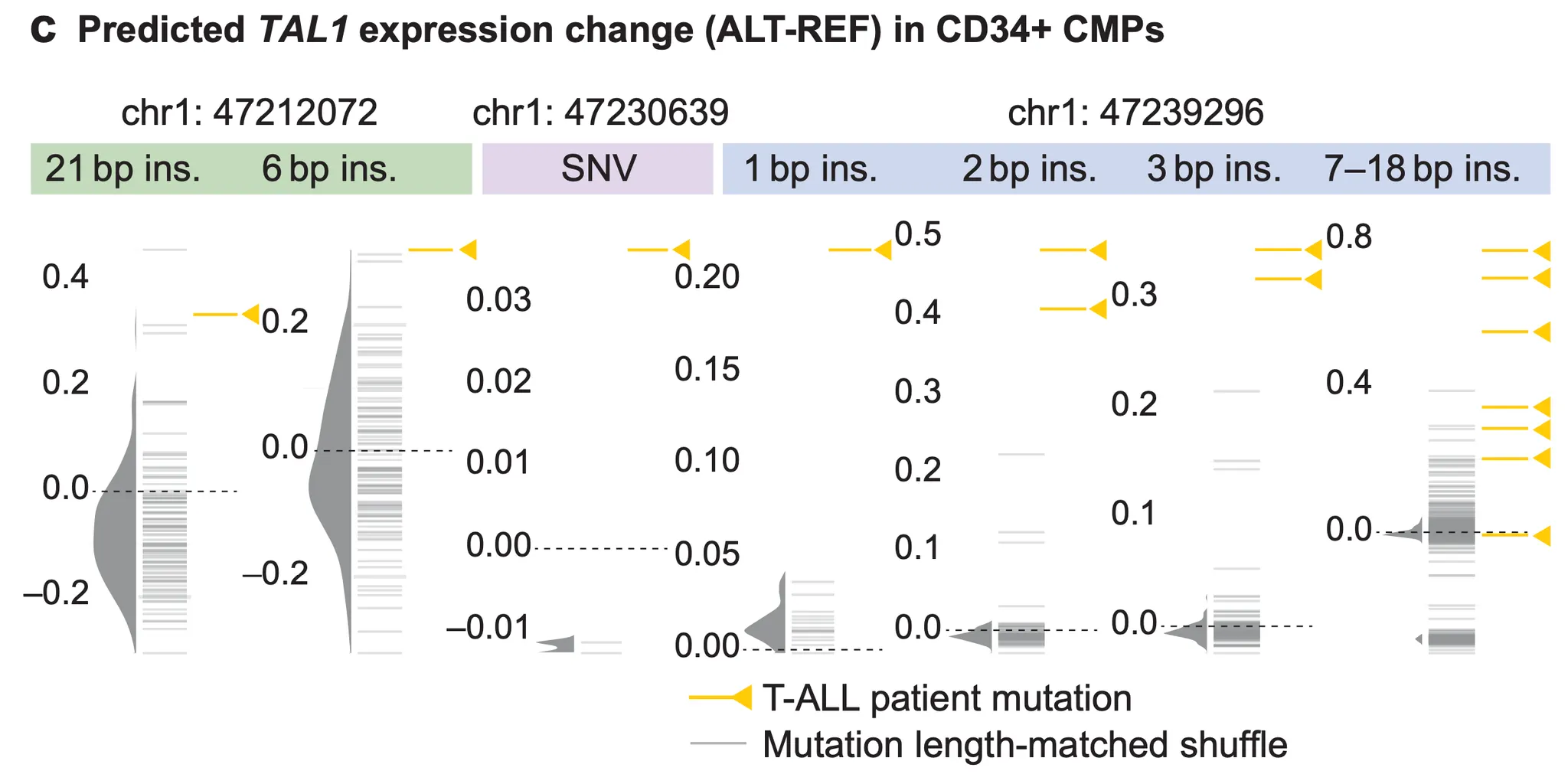
Fig. 6c - T-ALL 환자 변이와 대조군 변이의 TAL1 유전자 발현량 변화량 예측 비교.
위 그림의 y축은 예측된 TAL1 유전자 발현량의 변화량을 나타냅니다. 위로 올라갈수록 “발현량이 커질 것”으로 예측되는 겁니다. 노란색 화살표로 표시된 발현량 수준은 T-ALL 환자들에게서 나타나는 변이에 대한 발현량 예측값이고, 회색의 분포는 “같은 길이의 무작위 변이”들에 대한 발현량 예측 값 분포입니다. T-ALL 환자들에게서 나타나는 변이들은 TAL1 발현량을 증가시킨다고 알려져 있으므로, AlphaGenome 예측이 우리의 예상을 벗어나지 않는, 그럴듯한 예측임을 알 수 있겠군요!
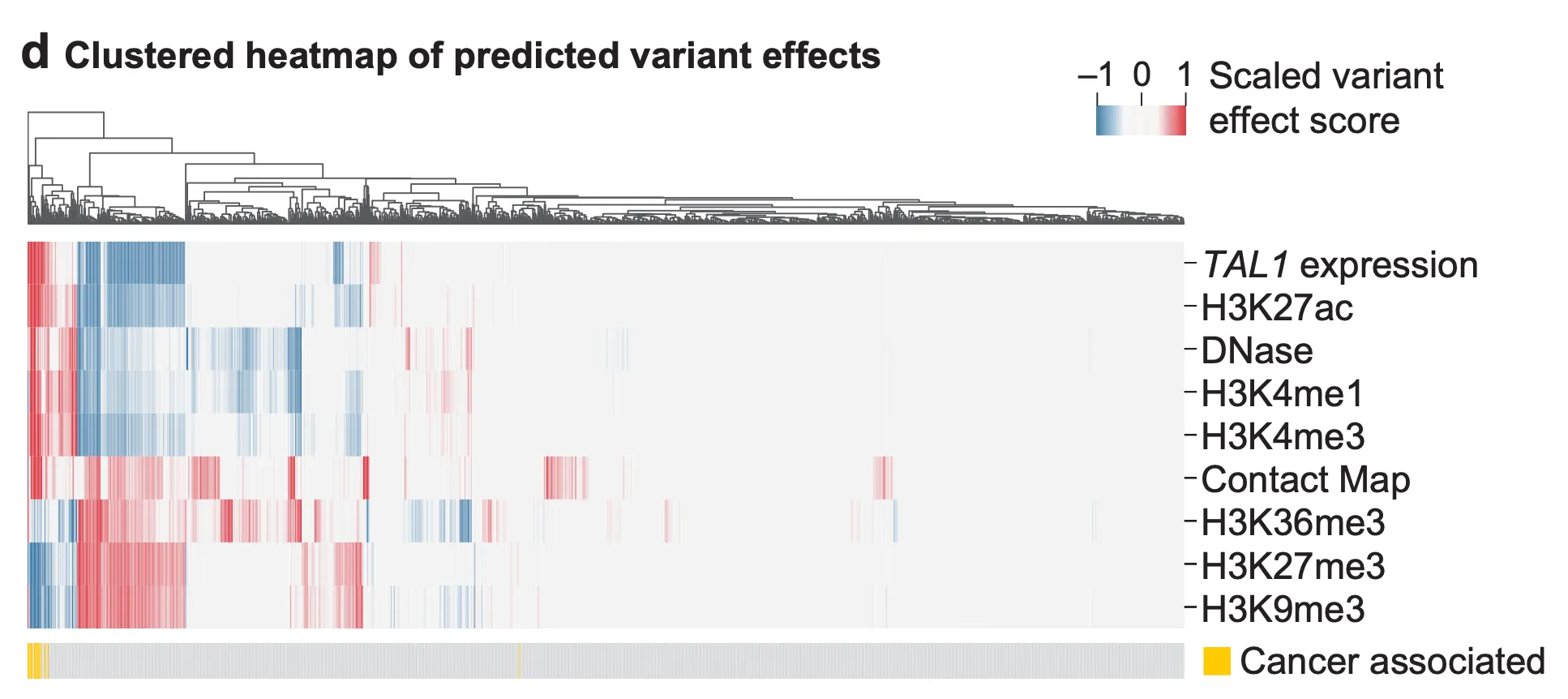
Fig. 6d - 변이에 의한 시그널 변화량 패턴의 군집화 결과.
AlphaGenome의 TAL1 과발현 예측을 더욱 뒷받침하기 위해, 저자들은 변이에 의해 나타나는 track들의 패턴을 군집화(clustering)하여 보여줍니다. 위 그림의 가장 아래 가로줄에서, 노란색으로 표시된 T-ALL 환자들에게서 나타나는 변이들은 잘 군집화 되어 있음을 알 수 있습니다. 즉, 다양한 변이들의 유전체의 기능적 변화 패턴을 매우 유사하게 예측했다는 점에서 AlphaGenome 예측의 신빙성을 높여준다고 볼 수 있겠습니다.
지금까지의 결과를 정리하면, AlphaGenome이 T-ALL 호발 변이들이 유사한 기능적 변화에 의해 공통적으로 TAL1 과발현을 유도함을 멀티-모달 분석을 통해 알 수 있었습니다. 그렇다면 보다 직접적으로, 왜 이러한 변이가 네오-인핸서의 활성화로 이어지는지, 보다 직접적인 서열 수준에서의 해석이 가능할까요?
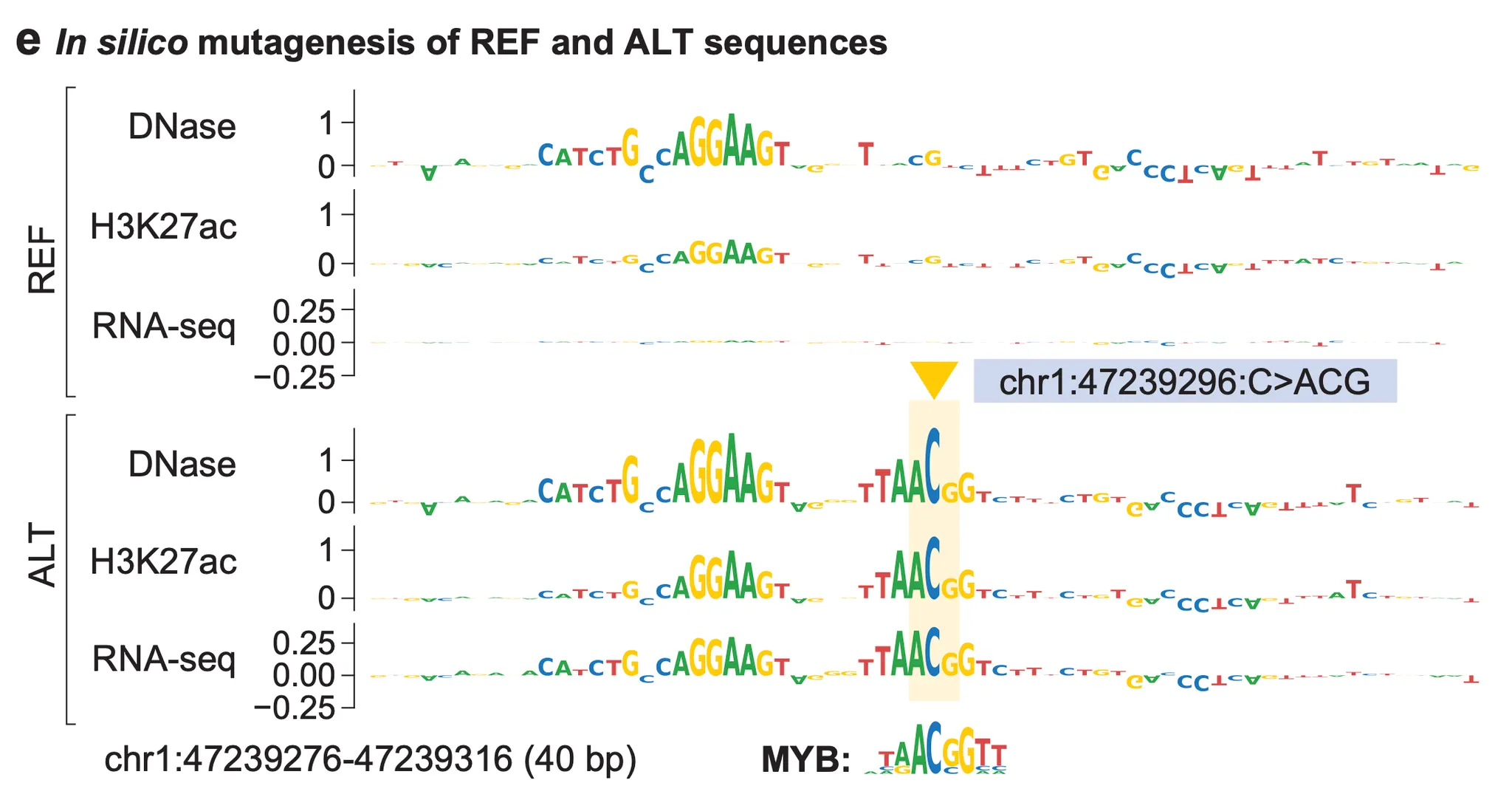
Fig. 6e - 참조 서열(REF)과 변이 서열(ALT)에 대한 in silico mutagenesis 실험 결과.
위 그림은 변이가 없는 참조 서열(reference; REF)과 변이가 있는 서열(alternative; ALT)에서 모두 ISM 실험을 수행한 결과입니다. 앞서 살펴보았던 ISM 실험 결과의 해석 방법을 참고해서 위 그림을 해석해 보면, “chr1:47239296:C>ACG 변이가 생기자 해당 ACG 부분이 DNase, H3K27ac 및 유전자 발현량 증가에 유의미하게 많이 기여하게 되었다”고 볼 수 있겠네요. 흥미롭게도 이 지역에 ACG 염기 삽입에 의해 새로 만들어진 서열은 MYB라는 전사 인자 결합 서열과 일치하며, 이는 과거 Mansour et al. 의해 이미 알려진 사실입니다.
지금까지 AlphaGenome이 유전체의 문법을 얼마나 잘 이해하고 있는지를 Variant effect prediction이라는 문제에 대한 성능 평가를 통해 확인해 보았습니다. 많은 non-coding variant의 기능적인 역할에 대해, 다양한 모달리티에 걸친 흥미로운 해석이 가능함을 알 수 있었네요. 그렇다면 어떻게 이렇게 좋은 성능을 갖는 모델을 만들 수 있었는지, 모델 개발 관점에서 그 비법에 대한 힌트를 얻을 수 있을지 알아보겠습니다.
AlphaGenome 모델 구조
지금까지 AlphaGenome 모델의 활용 가치를 알아보았으니, 이제 AlphaGenome 모델의 아키텍쳐와 학습 방법에 대해 자세히 알아봅시다.
모델 학습 방법
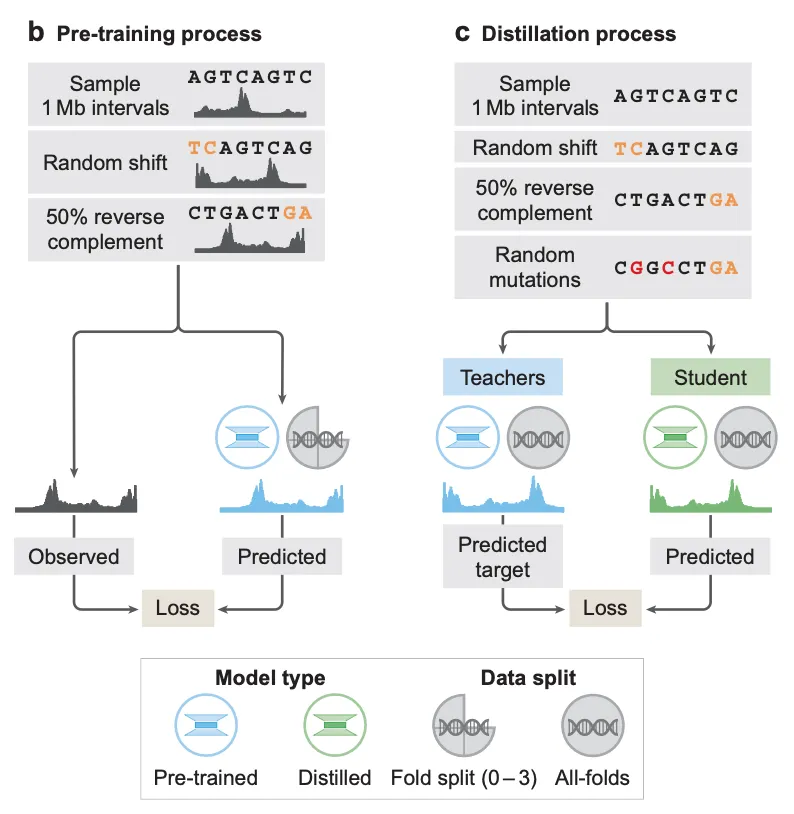
AlphaGenome은 비교적 색다른 2단계 학습 방법을 취합니다.
1단계를 사전학습(pretraining) 단계라고 부르며, 일반적인 인공지능 모델 학습과 크게 다를 바 없이 입력 DNA 서열이 주어졌을 때 genomic track을 예측하는 학습을 수행합니다. 확인해두어야 할 것은, 1단계 사전학습에서는 2종류의 모델 학습을 수행한다는 것인데, (1) 첫째로는 유전체에 존재하는 전체 genomic interval의 3/4만 가지고 학습한 다음 나머지 1/4로 성능을 평가하는 cross-validation scheme의 학습과, (2) 유전체 전체(4/4)를 가지고 수행하는 학습입니다. 전자의 경우, AlphaGenome 모델 아키텍쳐 자체의 일반화(generalization) 성능을 보고자 함입니다. 즉, 학습 때 사용되지 않았던 genomic interval에 대해서도 예측을 잘 하는지 확인하는 학습이라고 보시면 됩니다. 반면 후자의 방법으로 학습된 “all-folds” 모델의 경우, 뒤에 설명할 증류(distillation) 학습에서 teacher 모델로써 활용되게 됩니다.
2단계는 증류 학습입니다. 앞서 유전체 전체를 가지고 학습한 “all-folds” 모델을 여러 개(64개) 학습시켜둔 뒤, 학습에 사용되는 genomic interval instance에 대해서 여러 teacher 모델의 예측을 만들고 student 모델이 teacher 모델의 예측을 모방하도록 학습합니다. (실제 구현에서는 하나의 genomic interval에 대해서 모든 teacher로 하여금 예측을 만들어내도록 하지는 않고, 각 teacher 모델 별로 무작위로 샘플링된 genomic interval에 대한 예측을 만들어내도록 하여 학습을 진행합니다.)

증류 학습이란?
일반적으로 증류 학습(Distillation)은 크고 복잡한 모델(teacher 모델)이 학습한 지식을 작고 효율적인 모델(student 모델)로 전달하는 기법입니다. 교사 모델은 높은 정확도를 보이지만 연산 비용이 많이 드는 반면, 학생 모델은 적은 리소스로도 교사 모델과 유사한 성능을 낼 수 있도록 훈련됩니다.
단, AlphaGenome에서처럼 teacher 모델과 student 모델의 크기 차이가 없어도 넓은 의미에서 증류 학습이라고 부르기도 합니다. 여러 개의 teacher 모델 앙상블을 통해서 달성할 수 있었던 성능을 단일 student 모델로 달성할 수 있기에 증류 학습의 목적 중 하나인 리소스 효율성을 달성할 수 있다고 이해하면 되겠습니다.
모델 구조 및 학습에서, 어떤 부분이 효과적이었을까?

요약
•
스플라이싱 예측, 염색질 접근성 예측 등 높은 해상도가 요구되는 타겟에 대해서 높은 성능을 달성하기 위해서는 단일염기(1bp) 수준의 높은 해상도 데이터로 학습하는 것이 필수적
•
1Mb 수준의 큰 서열 윈도우로 학습하고, 마찬가지로 1Mb 서열 윈도우로 추론하는 것이 가장 높은 성능을 보임
•
Pretrained model의 ensemble 결과를 예측하도록 하는 distillation 학습을 통해 향상된 성능을 가지는 단일 모델 확보가 가능함
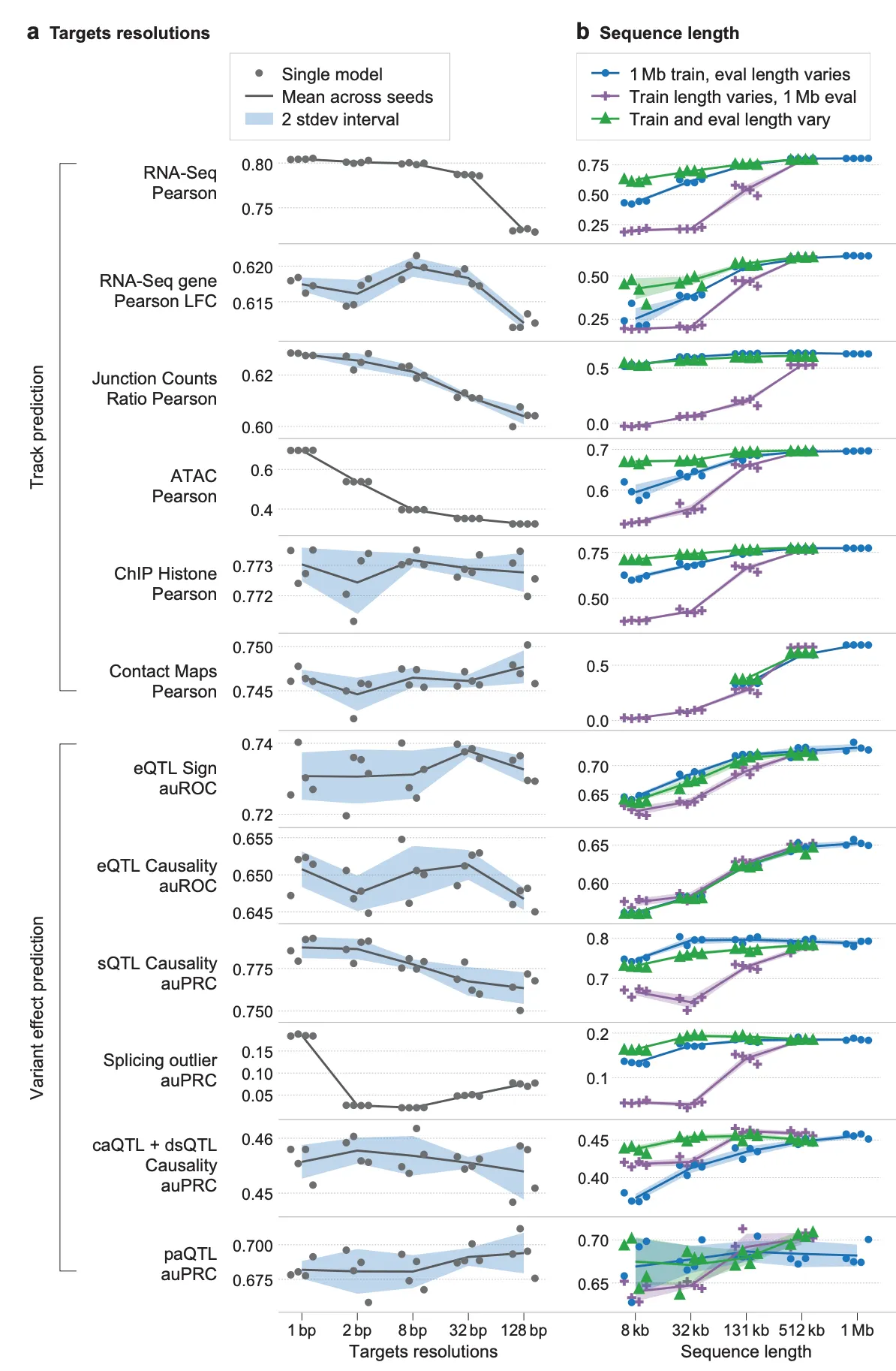
Target track의 해상도(resolution, 왼쪽)와, 학습/추론 시 사용하는 서열 윈도우의 크기(오른쪽)와 모델 성능 사이의 관계.
가장 먼저, 모델 학습에 사용하는 target track의 해상도(resolution)가 모델 성능에 미치는 영향을 확인해본 결과(위의 그림, 왼쪽)를 요약하면 아래와 같습니다.
Target track 해상도의 영향
•
가장 좋은 해상도인 1bp로 학습하는 것이 일관되게 좋은 성능을 보여줌.
•
특히 splicing이나 ATAC-seq signal과 같이 task 자체가 높은 해상도를 필요로 하는 경우 경향이 두드러짐.
•
반면, 히스톤 ChIP-seq 이나 contact map 예측과 같이 상대적으로 task 자체의 해상도가 낮은 경우 (즉, 염기 단위에서 수행되는 기능이 아니라, 일정 범위의 유전체 지역에서 일어나는 task인 경우), 학습 해상도에 따른 성능 영향이 크지 않음.
다음으로, 학습/추론 시 사용하는 서열의 길이에 따른 성능 차이(위의 그림, 오른쪽)를 요약하면 다음과 같습니다. 그림에서 파란색 선은 1Mb (1,000,000bp) 길이의 서열로 학습한 모델을 x축에 나타난 길이의 서열로 추론한 결과이며, 반대로 보라색 선은 x축에 나타난 길이의 서열로 학습한 모델을 1Mb 길이의 서열로 추론한 결과입니다. 초록색 선은 학습/추론 시의 서열 길이가 모두 x축 값과 일치한다고 보시면 됩니다.
학습/추론 시 서열 길이의 영향
•
1Mb 서열로 학습하고, 1Mb 서열로 추론하는 경우 대체로 가장 좋은 성능을 보임.
•
(보라색 선) 추론을 아무리 긴 서열로 하더라도 (1Mb), 애초에 긴 서열을 이용하여 학습된 모델이 짧은 서열로 학습된 모델보다 유의미하게 좋음.
•
(파란색 선) 추론 시의 서열 context의 길이도 길수록 좋음.
•
(초록색 선과 파란색 선 비교) 각 추론 서열 길이에 맞춤형으로 학습된 초록색 결과와, 1Mb 서열로 학습한 모델을 다양한 추론 서열 길이로 평가한 파란색 결과를 비교해보면, 1Mb 서열로 학습한 모델이 맞춤형-길이 모델에 아주 많이 뒤처지지는 않는 모습임. 이를 이용하여, 1Mb 서열로 학습된 모델을 가지고 짧은 서열로 추론하는 식으로 모델 배포 시의 추론 속도에서의 이점을 가져갈 수 있음.
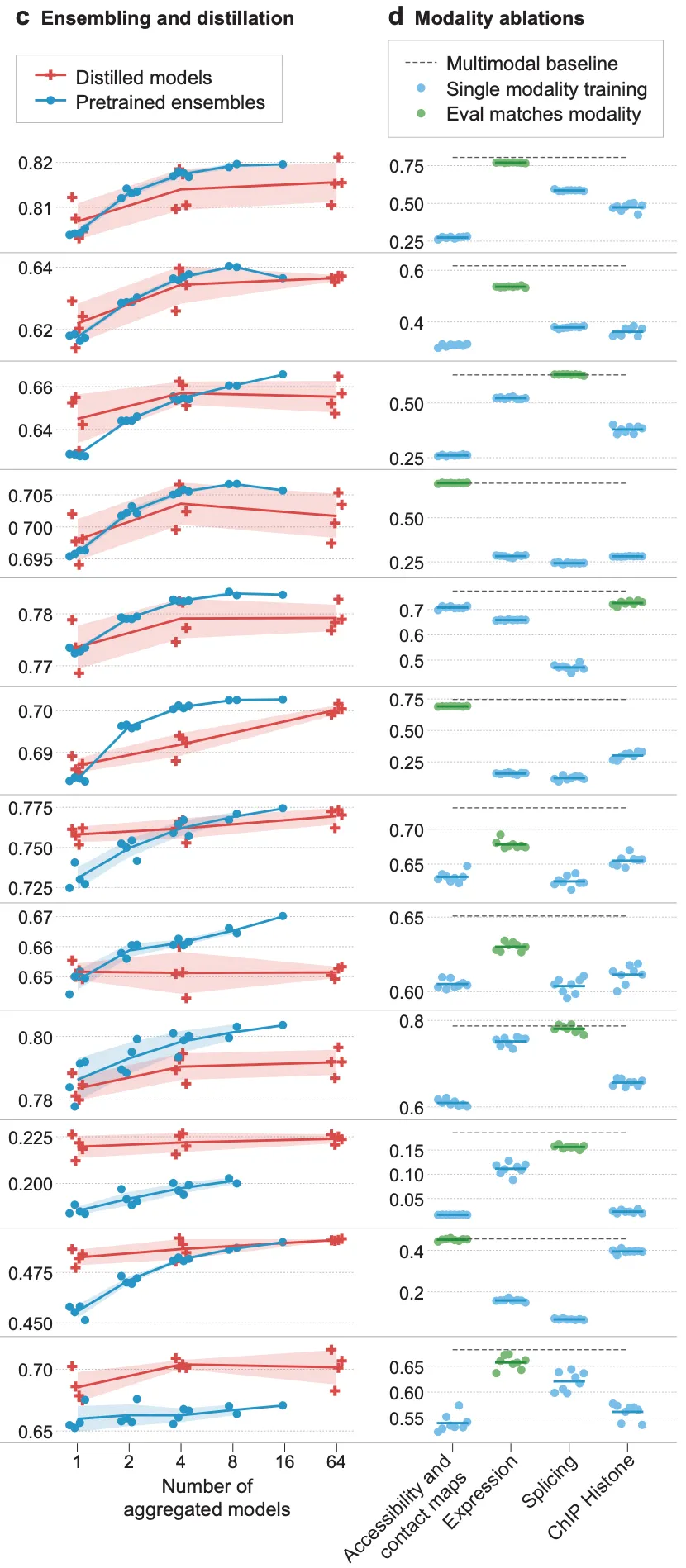
증류(distillation) 방법과, 앙상블 방법 사이의 성능 차이에 관한 ablation study 결과(위의 그림, 왼쪽)도 보여주고 있네요. 요지는, 여러 teacher 모델의 distillation을 통해 얻은 1개의 모델이, 여러 teacher 모델 예측 값의 앙상블 자체와 어느 정도 비슷한 수준의 성능을 달성할 수 있다는 점입니다. 그림의 빨간색 십자가들은 왼쪽부터 1개, 4개, 64개의 teacher 모델을 distillation에 사용하여 얻은 단일 모델의 예측 값들이고, 파란색 점들은 teacher 모델의 예측 값 앙상블 성능입니다. Distillation 결과들이 항상 teacher 모델 성능을 뛰어넘는 것은 아니지만, teacher 모델 1개의 성능(가장 왼쪽의 파란색 점들)과 비교해본다면 단일 모델로서는 확실히 더 향상된 성능(가장 오른쪽의 빨간색 십자가들)을 보인다는 것을 알 수 있습니다.
끝으로, 여러 task를 동시에 학습하는 멀티모달(multimodal) 학습이 성능에 어떤 이점을 가져다주는지 보여주고 있습니다(위의 그림, 오른쪽). 결과를 요약하면 아래와 같습니다.
멀티모달 학습의 이점
•
일반적으로, 멀티모달 학습 모델이 개별 모달리티에 대해 학습된 모델의 성능을 능가함. 여러 task에 걸친 shared representation을 학습하는 것의 이점을 암시함.
•
물론 task-dependent한 경우도 있음.
◦
예를 들어, caQTL 예측과 같은 경우, 직접적으로 관련된 acessibility 데이터만 가지고 학습해도 multi-modal 학습 결과와 같은 성능을 보임. 즉, 이 task에 한해서는 멀티모달 학습의 이점이 크게 없다고 볼 수 있음.
◦
반대로 eQTL 예측의 경우, 직접적으로 관련된 expression 데이터만 가지고 학습하는 것보다 다른 모달리티를 함께 사용하여 학습하는 편이 성능이 더 좋음. 즉, 멀티모달 학습의 이점이 있다고 볼 수 있음.
AlphaGenome 학습 세부사항
AlphaGenome 논문의 Method에는 꽤 방대한 분량으로 데이터 전처리 방법이 설명되어 있습니다. 다양한 오믹스 데이터를 전처리하기 위해서는 각 오믹스에 특화된 데이터베이스 및 소프트웨어의 활용과, 적절한 파라미터 값 선정이 중요한데요, 다음 포스팅에서는 AlphaGenome 논문에서 어떤 방식으로 각 오믹스 데이터를 취합하고, 전처리했는지 알아보면서 그들의 노하우를 들여다보도록 하겠습니다.