SeedFold 논문 리뷰
들어가며
AlphaFold3 이후 biomolecular structure prediction의 경쟁은 단순히 protein monomer를 더 잘 맞히는 문제에서 벗어났습니다. 이제는 protein-protein complex, antibody-antigen interface, protein-ligand pose, protein-RNA/DNA interaction까지 하나의 all-atom framework 안에서 얼마나 안정적으로 예측할 수 있는지가 중요해졌습니다.
SeedFold는 ByteDance Seed가 발표한 2025년 arXiv preprint입니다. 이 글에서는 SeedFold를 binder generator나 antibody design paper가 아니라, AF3-style biomolecular structure predictor를 어떻게 scale할 것인가를 다룬 architecture/data-scaling paper관점에서 살펴보겠습니다.
핵심 메시지는 세 가지입니다. 첫째, Pairformer는 depth보다 width가 병목일 수 있습니다. 둘째, triangular attention의 cost를 줄이기 위해 linear triangular attention을 넣을 수 있습니다. 셋째, AFDB/MGnify 기반 distillation data가 AF3-style model scaling의 중요한 재료가 됩니다.
AF3-style predictor에서 병목 찾기
AlphaFold3 계열 모델은 sequence와 biomolecular context를 token/pair representation으로 바꾸고, Pairformer가 interaction representation을 업데이트한 뒤, diffusion-style structure module이 coordinates를 생성하는 구조로 이해할 수 있습니다. 이 구조에서 Pairformer는 residue, ligand atom, nucleic acid token 사이의 pairwise relationship을 담는 핵심 trunk입니다.
SeedFold는 이 trunk의 capacity가 어디에서 막히는지를 묻습니다. 단순히 layer를 더 깊게 쌓으면 되는지, structure module을 더 크게 만들면 되는지, 아니면 pair representation의 width를 키우는 편이 더 효율적인지 비교합니다.
이 질문은 practical합니다. Biomolecular complex prediction은 monomer prediction보다 pairwise interaction이 훨씬 중요합니다. Antibody-antigen interface, ligand pocket, protein-RNA contact는 모두 “두 token 사이의 관계”에 민감합니다. SeedFold가 Pairformer width에 주목하는 이유입니다.
Pairformer width scaling
SeedFold의 첫 번째 claim은 folding model capacity가 Pairformer layer 수보다 pair representation hidden dimension에 의해 bottleneck될 수 있다는 것입니다. 논문은 Base, Medium, Large model과 Deep Pairformer, Deep Structure Module variant를 비교합니다.
Base는 pair dim 128, MSA dim 64, Pairformer 48 layers, Structure Module 24 layers로 432M parameters입니다. Medium은 pair dim 256 / MSA dim 128로 533M parameters입니다. Large는 pair dim 512 / MSA dim 256으로 923M parameters입니다. Deep Pairformer는 Pairformer layer를 96으로 늘리고, Deep Structure Module은 structure layers를 48로 늘립니다.
논문 해석은 width scaling이 global RMSD와 intra-protein lDDT를 가장 잘 개선한다는 쪽입니다. Base에서 Medium으로 갈 때 gain이 크고, Medium에서 Large로 갈 때는 diminishing return이 보입니다. Deep Pairformer는 recycling 때문에 이미 effective depth가 커서 gain이 제한적이고, structure module은 pair representation을 coordinate로 translate하는 역할에 가까워 scaling 효과가 작다고 봅니다.
이 결과는 AF3-style model을 해석하는 데 유용합니다. Folding model의 성능을 올리는 길이 무조건 더 깊은 network가 아니라, pair representation의 폭과 그 안에 담기는 interaction capacity일 수 있다는 뜻입니다.
Triangular attention의 비용
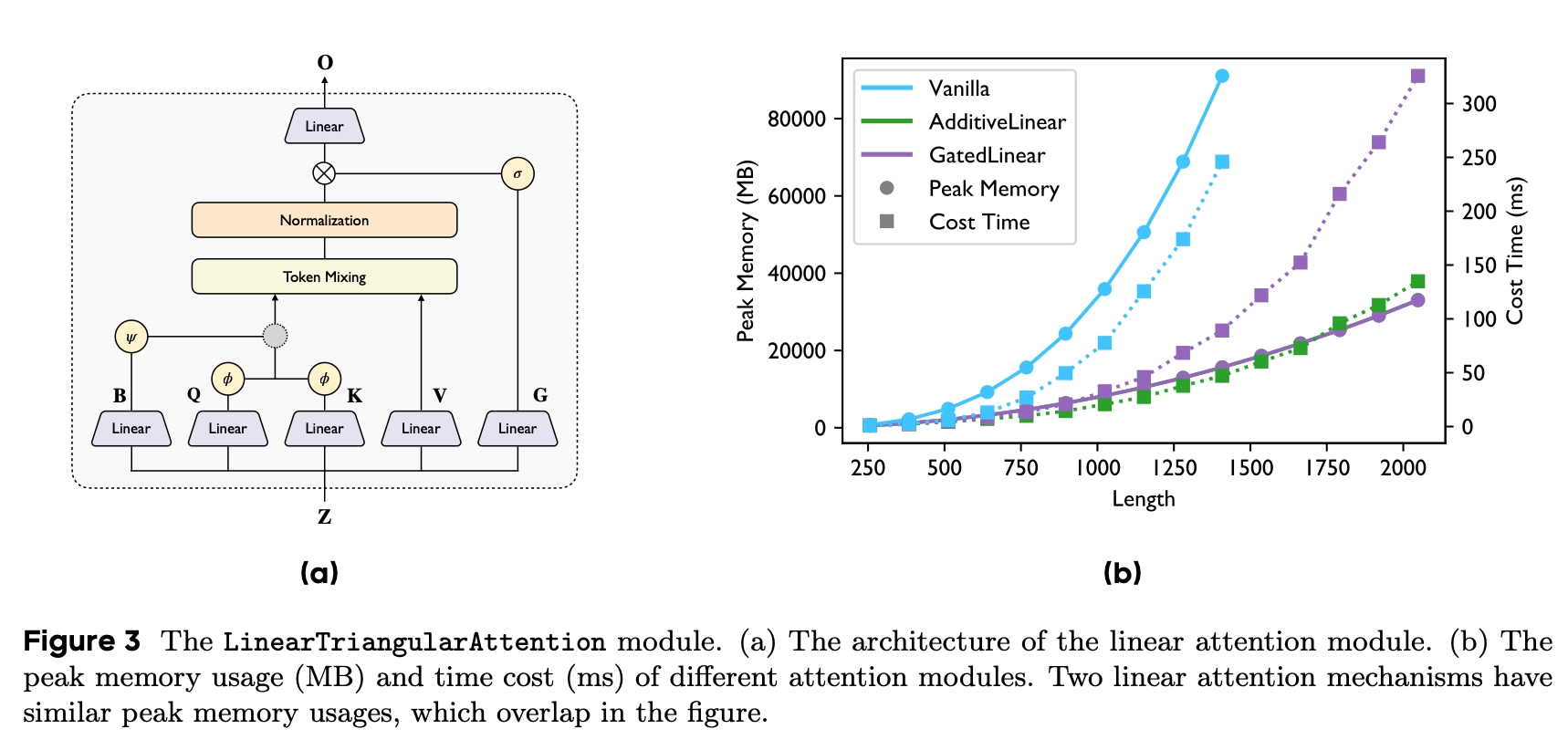
Pairformer에서 중요한 연산 중 하나는 triangular operation입니다. Protein structure에서는 residue i-j, j-k, i-k 관계가 함께 움직입니다. Triangle update나 triangular attention은 이런 3-body relational consistency를 다루는 장치입니다.
문제는 비용입니다. Vanilla triangular attention은 row/column마다 token-token score matrix를 만들기 때문에 token length에 대해 cubic하게 비싸질 수 있습니다. Biomolecular complex는 ligand, nucleic acid, multimer chain까지 들어가면 token 수가 커지고, triangular attention cost가 scale-up의 병목이 됩니다.
SeedFold는 이 비용을 줄이기 위해 Linear Triangular Attention을 제안합니다. 일반 softmax attention을 feature map 기반 linear attention으로 바꾸되, folding model에서 중요한 triangular form과 pair bias를 최대한 유지하려는 접근입니다.
Additive와 Gated linear triangular attention
논문은 두 variant를 제시합니다. AdditiveLinearTriAtt는 AlphaFold-style pair bias를 additive하게 넣어 기존 attention bias의 장점을 유지하려는 방식입니다. GatedLinearTriAtt는 pair bias를 gating signal처럼 사용해 QK correlation의 information flow를 조절합니다.
SeedFold-Linear는 384-width model에 GatedLinearTriAtt를 넣은 variant입니다. 논문은 GatedLinearTriAtt가 DNA/RNA-related tasks와 antibody/protein-ligand interface comparison에서 약하지만 일관된 장점을 보였기 때문에 기본 linear variant로 선택했다고 설명합니다.
여기서 중요한 점은 linear attention이 “항상 더 좋은 모델”이라는 뜻은 아니라는 것입니다. SeedFold-Linear는 protein-ligand와 protein-DNA 쪽에서 강한 수치를 보이지만, vanilla SeedFold가 antibody-antigen과 protein-RNA 쪽에서 더 강합니다. Linear triangular attention은 scaling cost를 줄이는 promising direction이지만, task별 trade-off를 같이 봐야 합니다.
Distillation data가 커지는 방식
SeedFold의 또 다른 축은 large-scale distillation입니다. Experimental PDB data만으로 general-purpose AF3-style structure module을 안정적으로 학습하기 어렵다고 보고, AF2/OpenFold-derived predicted structures를 대규모로 사용합니다.
Training set은 PDB 180,540 samples, AFDB 3,326,991 samples, MGnify 23,075,211 samples로 구성됩니다. Sampling weight는 PDB 0.50, AFDB 0.08, MGnify 0.42입니다. 논문은 total training set을 26.5M samples로 설명하고, experimental dataset 0.18M의 147배 규모라고 말합니다.
이 규모는 SeedFold의 강한 부분입니다. AFDB는 UniProt-derived AF2-predicted structures에 가깝고, MGnify는 metagenomic sequence diversity와 longer protein distribution을 제공합니다. 특히 MGnify 23M samples 중 AFDB cluster에 assign되는 것이 약 2M뿐이었다는 점을 들어, sequence diversity를 강조합니다.
다만 distillation data는 experimental ground truth가 아닙니다. AF2/OpenFold-derived prediction layer입니다. 학습에는 강한 prior가 될 수 있지만, benchmark claim을 읽을 때 pseudo-label dependency를 분리해두면 충분합니다.
Training setup과 precision detail
SeedFold는 two-stage training을 사용합니다. Stage 1은 crop size 384 tokens, diffusion batch size 64, 60k iterations입니다. Stage 2는 crop size 640 tokens, diffusion batch size 32, additional 40k iterations입니다.
Batch size는 256, optimizer는 AdamW, maximum learning rate는 0.0018입니다. MSA는 10% probability로 drop하고, monomer distillation data는 50% ratio로 sample합니다.
Precision detail도 흥미롭습니다. MSA module과 Pairformer는 bfloat16을 쓰지만, coordinate-space에서 작동하는 Structure Module은 float32로 유지합니다. 논문은 Structure Module을 bfloat16으로 돌리면 local distance metric인 lDDT가 크게 떨어지고, global DockQ는 상대적으로 덜 변한다고 적습니다. Coordinate-level local accuracy에는 numerical precision이 민감하다는 뜻입니다.
Scaling instability도 기록됩니다. Pairformer width가 256을 넘으면 early training에서 gradient norm explosion과 loss collapse가 나타났고, warmup을 1000에서 3000으로 늘리고 512-width large model에는 lower learning rate를 사용합니다. Scale-up은 단순히 parameter를 늘리는 문제가 아니라 optimization recipe와 함께 움직입니다.
FoldBench benchmark
SeedFold는 FoldBench 1,522 biological assemblies를 사용합니다. FoldBench는 2023년 1월 13일 이후 deposited structures에서 구성되며, protein/RNA/DNA monomers와 protein-protein, antibody-antigen, protein-ligand, protein-RNA, protein-DNA, protein-peptide interfaces를 포함합니다.
Evaluation setting은 5 random seeds × 5 diffusion samples, 10 recycling steps, model ranking score로 best prediction을 고르는 방식입니다. 이 sampling/ranking budget은 benchmark number의 일부입니다. 구조 예측 모델 비교에서는 model architecture뿐 아니라 seed 수, diffusion samples, recycling, ranking score가 모두 영향을 줍니다.
Main result에서 SeedFold는 monomer lDDT 0.8889, protein-protein DockQ success 74.03%, antibody-antigen DockQ success 53.21%, protein-RNA DockQ success 65.31%를 보고합니다. SeedFold-Linear는 protein-protein 74.14%, protein-ligand 66.48%, protein-DNA 76.00%에서 강합니다.
AlphaFold3와 비교하면 SeedFold는 antibody-antigen에서 53.21%로 AF3 47.90%보다 높고, protein-RNA에서도 65.31%로 AF3 62.32%보다 높습니다. Protein-ligand에서는 SeedFold-Linear 66.48%가 AF3 64.90%보다 높습니다. 반면 protein-DNA와 RNA lDDT에서는 AF3가 더 높고, DNA lDDT는 Protenix-0.5가 가장 높습니다.
Antibody-antigen과 protein-ligand result의 의미
SeedFold의 headline 중 하나는 antibody-antigen DockQ success입니다. SeedFold 53.21%는 Boltz-1 33.54%, Chai-1 23.64%, Protenix-0.5 41.00%, AlphaFold3 47.90%보다 높게 보고됩니다. Antibody-antigen interface prediction은 antibody design pipeline에서 후보 pose를 평가하는 데 중요하므로, 이 수치는 downstream filtering 관점에서 눈에 띕니다.
Protein-ligand에서는 SeedFold-Linear가 66.48% success rate를 보고합니다. AF3 64.90%, Boltz-1 55.04%, Chai-1 51.23%, Protenix-0.5 62.30%와 비교되는 수치입니다. Ligand pose prediction과 pocket plausibility는 small-molecule design, docking, affinity modeling의 upstream layer가 될 수 있습니다.
하지만 이 결과들은 structure-prediction benchmark입니다. Antibody-antigen DockQ가 높다는 말은 antibody가 실제로 binding한다는 뜻이 아니고, protein-ligand SR이 높다는 말도 affinity나 activity를 보여주지 않습니다. SeedFold는 evaluator/filter infrastructure 후보로 이해해야지, wet-lab design success로 이해하면 안 됩니다.
Linear variant의 위치
SeedFold-Linear는 흥미로운 variant입니다. GatedLinearTriAtt를 넣어 triangular attention cost를 줄이면서, protein-ligand와 protein-DNA에서 강한 성능을 보입니다. 논문은 vanilla triangular attention과 linear variants가 대부분 task에서 비슷하고, GatedLinearTriAtt가 nucleic-acid-related tasks에서 우세하다고 보고합니다.
다만 SeedFold-Linear는 384-width model입니다. 논문은 resource limitation과 convergence issue 가능성 때문에 linear-attention model을 더 키우지 않았다고 적습니다. 따라서 SeedFold-Linear의 protein-ligand result는 promising하지만, 512-width vanilla SeedFold와 완전히 같은 scaling state에서 비교된 것은 아닙니다.
이 점은 중요합니다. Linear attention은 cost를 줄이는 방향이고, width scaling은 capacity를 늘리는 방향입니다. 두 축이 아직 완전히 결합된 최종 답이라기보다, SeedFold 논문은 “어디를 키워야 하는가”와 “어디를 줄일 수 있는가”를 함께 제시한 것으로 보는 편이 맞습니다.
Figure와 ablation 읽기
Scaling strategy figure는 width scaling이 depth scaling보다 global RMSD와 intra-protein lDDT에서 더 좋은 방향임을 보여줍니다. 정확한 numeric table보다 방향성이 중요합니다. Pairformer의 폭이 interaction representation capacity를 좌우한다는 해석입니다.
LinearTriangularAttention figure와 kernel benchmark는 memory/time cost를 다룹니다. Folding model에서 triangular attention을 linearize하려는 시도가 단순 speed trick이 아니라, long token complex를 다루기 위한 scaling strategy라는 점을 보여줍니다.
Monomer distillation removal ablation도 중요합니다. 47,612-step checkpoint에서 distillation data를 제거하고 continued training하면 intra-protein lDDT가 즉시 악화된다고 보고합니다. 이는 AFDB/MGnify distillation이 단순 data augmentation이 아니라 model quality 유지에 직접적인 역할을 한다는 evidence입니다.
Evidence layer를 분리해서 읽기
SeedFold의 evidence는 세 층으로 나눌 수 있습니다. 첫 번째는 architecture scaling evidence입니다. Pairformer width scaling, deep Pairformer, deep Structure Module 비교가 여기에 들어갑니다.
두 번째는 efficiency/scaling evidence입니다. Linear triangular attention, GatedLinearTriAtt, kernel benchmark, memory/time cost가 이 층입니다.
세 번째는 structure-prediction benchmark evidence입니다. FoldBench monomer lDDT, DockQ success rate, protein-ligand success rate, nucleic-acid metrics가 여기에 들어갑니다. 이 층은 design filtering과 연결될 수 있지만, wet-lab validation은 아닙니다.
이 논문을 읽을 때의 guardrail
첫째, SeedFold는 structure prediction paper입니다. Binder generation, antibody design, ligand design wet-lab validation을 보고하는 논문이 아닙니다.
둘째, FoldBench numbers는 sampling budget과 ranking strategy를 포함한 benchmark result입니다. 5 seeds × 5 diffusion samples, 10 recycling steps라는 setting을 함께 읽어야 합니다.
셋째, training data의 대부분은 AFDB/MGnify-derived distillation입니다. Experimental PDB와 같은 evidence layer가 아닙니다.
넷째, SeedFold-Linear는 promising하지만 384-width variant입니다. 512-width vanilla SeedFold와 같은 capacity state에서 비교된 것은 아닙니다.
다섯째, 모든 task에서 우월한 모델이라고 말하면 과합니다. DNA lDDT는 Protenix-0.5, RNA lDDT는 AlphaFold3가 더 높습니다. SeedFold의 강점은 protein-related and interface tasks에서 두드러집니다.
평가: structure prediction infrastructure의 scaling recipe
SeedFold의 가치는 “AlphaFold3보다 좋은 모델”이라는 단순한 비교보다, AF3-style biomolecular predictor를 scale할 때 어디가 병목인지 보여주는 데 있습니다. Pairformer width, triangular attention cost, distillation data scale, precision/optimization recipe가 모두 성능에 연결됩니다.
특히 Pairformer width scaling은 중요한 메시지입니다. Biomolecular complex prediction에서는 pair representation이 interface reasoning의 중심입니다. Layer를 더 깊게 쌓는 것보다, pair representation이 더 많은 relational signal을 담도록 폭을 키우는 편이 효율적일 수 있다는 주장은 Protenix/Boltz/Chai 계열을 비교할 때 좋은 technical anchor가 됩니다.
다만 SeedFold는 아직 design validation paper가 아닙니다. Ab-Ag 53.21%, Prot-Lig 66.48% 같은 수치는 design pipeline의 evaluator/filter로서 기대감을 줍니다. 하지만 binding affinity, specificity, developability, wet-lab hit rate는 다른 evidence layer입니다. SeedFold는 구조 예측 infrastructure의 진화라는 관점에서 보는 편이 가장 정확합니다.
참고
- Paper: “SeedFold: Scaling Biomolecular Structure Prediction” - Authors: Yi Zhou, Chan Lu, Yiming Ma, Wei Qu, Fei Ye, Kexin Zhang, Lan Wang, Minrui Gui, Quanquan Gu - arXiv: https://arxiv.org/abs/2512.24354 - Project page: https://seedfold.github.io/ - Raw source: `raw/papers/SeedFold/seedfold.pdf` - Extracted source: `raw/papers/SeedFold/extracted/seedfold_source_text.txt`