Introduction
•
단백질 서열의 돌연변이 효과를 예측하는 unsupervised model은 신약 개발 및 병원성 돌연변이 예측에서 중요한 도구로 인기를 끌고 있다.
MSA 기반 방법론
•
그 중에서도 다중 서열 정렬(multiple sequence alignment, MSA) 기반의 방법론들이 좋은 성능을 보여왔는데, 여기서 MSA의 역할은 두 가지이다.

MSA의 역할
1.
Data acquisition tool: 관심 있는 단백질과 관련된 단백질들을 large DB에서 찾아준다
2.
Coordinate system: 정렬된 서열 상에서 특정 위치의 아미노산들의 비교를 가능하게 함
•
하지만 MSA는 치명적인 단점이 있다.
MSA의 단점
1.
잘 정립된 coordinate system 상에서의 아미노산 변이에 대해서만 예측이 가능하므로, insertion이나 deletion의 효과에 대해서는 예측이 어렵다.
2.
Proteome의 많은 부분은 정렬이 안되는 disordered region이다.
3.
Protein function이 특정 taxa에 국한되어 있다면 애초에 충분히 큰 MSA를 만들 수 없다.
4.
모델이 만들어진 MSA의 특성에 민감해질 수 있다.
a.
이러면 MSA를 만드는 parameter도 잘 조정해주어야 한다는 부담이 있음.
5.
서로 다른 data subset에 대해서 학습된 모델끼리 information sharing이 어렵다.
Language model (LM) 기반 방법론
•
MSA 기반 방법론을 해결하기 위해 LM 기반 방법론이 등장함.
◦
ESM-1v (Meier et al., 2021) → 대량의 non-aligned 단백질 서열로 학습
◦
MSA Transformer (Rao et al., 2021) → 대량의 aligned 단백질 서열로 학습
•
LM 방법론에도 한계는 있다.
LM 기반 방법론의 한계
1.
ESM-1v의 예를 들면, non-aligned 서열로 학습은 되지만 MSA 서열로 fine-tuning을 해야 성능이 쓸만해진다.
2.
Masked LM objective로 학습된 모델들은 full sequence의 likelihood를 계산하지 못한다.
a.
Mutation effect 예측에 있어서 어쩔 수 없이 heuristics의 도입이 필요해짐.
Tranception
•
Autoregressive transformer
•
대량의 non-aligned 단백질 서열로 학습한다. 학습 시에 MSA를 사용하지 않는다!
•
Inference-time retrieval 방법을 사용한다
•
단백질이 Shallow MSA를 갖더라도 성능이 좋다
모델 구조 및 데이터
Tranception attention
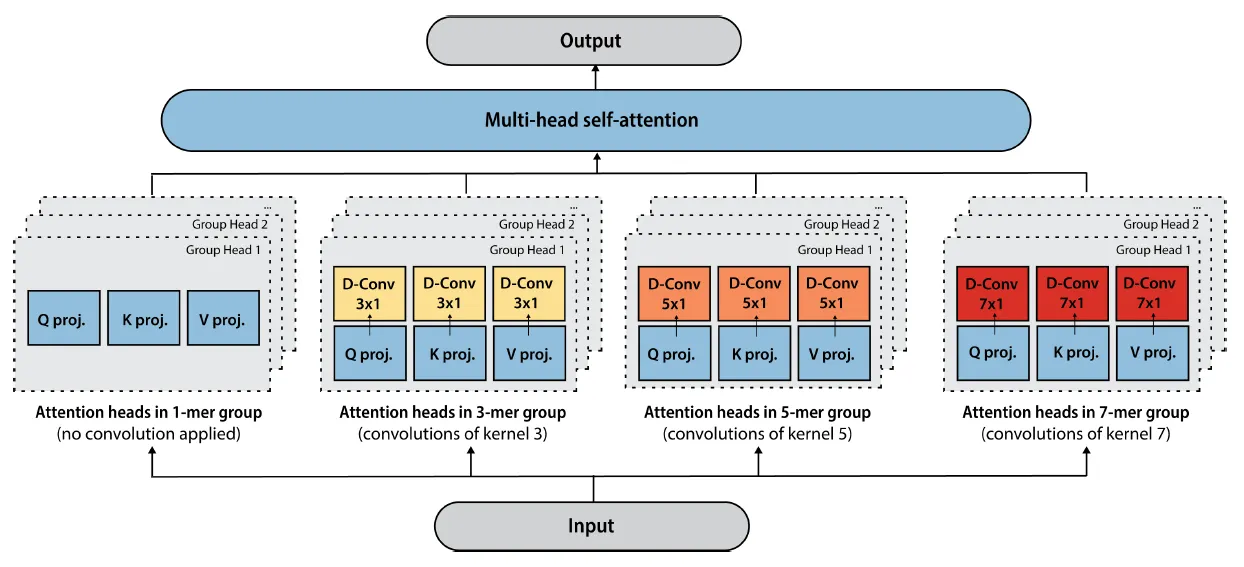
•
1-mer, 3-mer, 5-mer, 7-mer 단위로 convolution layer를 거친 후 attention을 수행한다.
◦
개별 attention head가 각 k-mer에 “specialized” 된다고 표현한다.
•
squared ReLU activation
•
D-conv = depthwise convolution
Grouped ALiBi position encoding
•
Learned position encoding이나 sinusoidal position encoding을 대신한다.
•
Attention with Linear Bias (ALiBi, 논문) 를 변형한 Grouped ALiBi를 사용한다. ALiBi는 기존의 position encoding을 사용하지 않고, 값에 두 position 간 거리에 비례하는 static한 bias를 더해주는 방법이다.
•
Grouped ALiBi도 기존 ALiBi와 같은 연산이지만, attention head group (k-mer group) 별로 따로따로 적용한다는 점만 다르다.
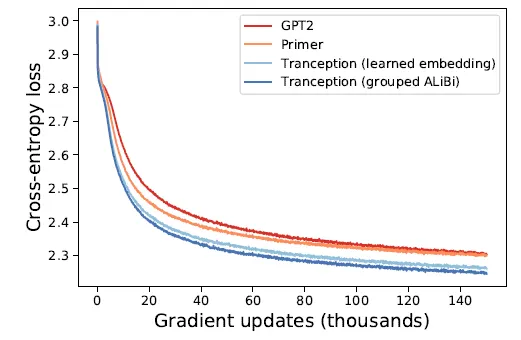
•
Grouped ALiBi를 사용하는 편이 learned embedding보다 파라미터 수도 적고, 수렴 속도도 빨라지는 것을 관찰했다.
Hyperparameters
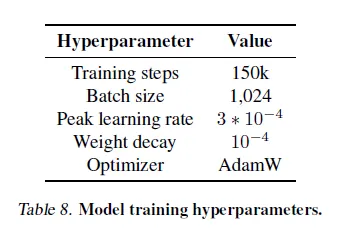
•
Learning rate schedule
◦
10k warmup steps to 3 * 10^-4
◦
linearly decreased until 150k steps
•
99% train (249 million seqs), 1% validation (2.5 million seqs)
•
UniRef50 cluster level에서 singleton인 단백질은 제거
•
Pyrrolysine (O)이나 Selenocysteine (U) 을 가진 단백질은 제거
•
2개 이상의 ‘X’ 아미노산이 있으면 제거
•
나머지 indeterminate ‘X, B, J, Z’ 아미노산은 다음과 같이 impute
◦
X는 20개 아미노산 중 하나로 무작위로 impute
◦
B는 D나 N으로
◦
J는 I나 L로
◦
Z는 E나 Q로
•
Validation에서는 indeterminate 아미노산을 가진 단백질 제거
Data processing and augmentations
•
UniRef100로 학습됐다.
◦
Clustering에 사용되는 similarity cutoff에 따라서 성능이 어떻게 변화하는지 봤더니, UniRef100 > UniRef90 > UniRef50 순으로 성능이 좋았다고 한다.
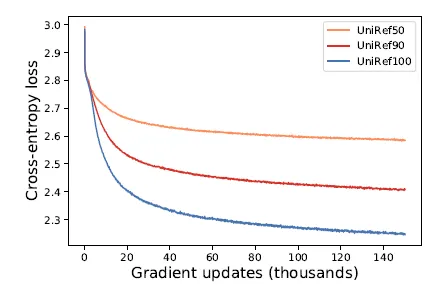
◦
아래는 preprocessing 이후의 dataset statistics. ~2억 5천개 단백질 서열로 학습.
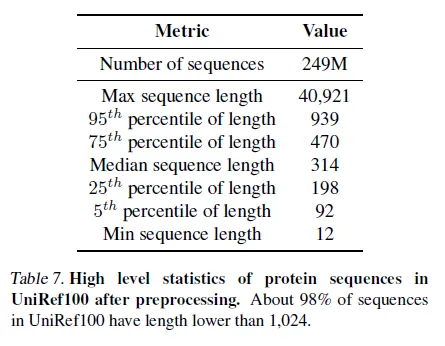
•
무작위로 단백질 서열을 역순으로 뒤집어서 학습에 사용.
◦
사실 단백질 서열은 방향성이 있어서 (N→C), 역순으로 뒤집으면 의미가 달라지지만 실험적으로 reverse sequence를 학습에 사용하면 성능이 좋아지는 것을 확인했다고 한다.
Scoring sequences for fitness prediction
•
Fitness prediction에 있어서 흔한 approach는, mutated sequence와 WT sequence의 likelihood ratio를 구하는 것
•
서열의 likelihood는 “어떤 position 이전의 서열들이 주어졌을 때 그 position의 아미노산의 존재 확률” 의 곱으로 나타난다.
Training
•
학습은 position 이전의 prefix 가 주어졌을 때, 의 확률을 예측하는 방식으로 진행된다.
◦
Autoregressive manner
Inference
MSA의 활용
•
어떤 단백질에 대한 MSA는 sequence space에서 이웃하고 있는 단백질들을 retrieve하여, query 단백질의 coordinate system으로 정렬하는 방법이라고 볼 수 있다.
•
MSA 상의 특정 위치에 나타나는 아미노산의 분포는 evolutionary constraint를 반영한다.
◦
MSA 상에 나타나는 아미노산 변이들은 fitness를 maintain하는 변이라고 볼 수 있다.
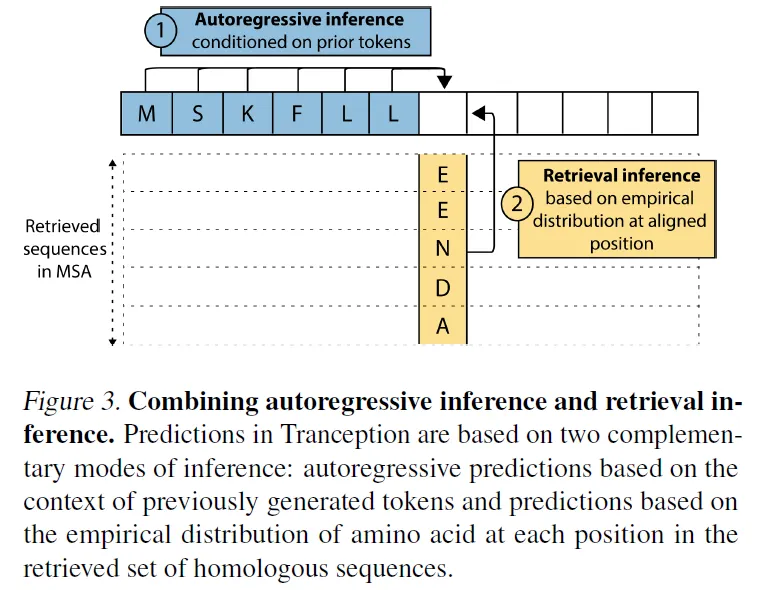
두 가지 방법의 inference (Autoregressive inference + retrieval inference)
Tranception은 두가지 inference 방법을 결합하여 서열의 likelihood를 예측한다.
1.
Autoregressive inference : 모델이 예측한 값들을 이용.
2.
Retrieval inference : MSA 상에 나타나는 empirical distribution을 이용
•
Insertion / Deletion의 경우에는?
◦
Insertion이 일어난 column에는 0을 채운다
◦
Deletion이 일어난 column은 삭제한다
•
Inference 시에 insertion이 일어난 column에 대해서는 retrieval inference를 수행하지 않고, autoregressive 모드만 사용한다.
•
Protein DB의 단백질들은 human sampling bias가 있기 때문에, sequence re-weighting을 수행한다. (Hopf et al., 2017, Neff로 normalize 하는 듯?)
•
Autoregressive 및 retrieval inference 시의 log likelihood를 각각 라고 하고, 전체 likelihood는 둘의 가중평균을 사용한다.
•
Autoregressive probability의 특성을 활용하면 결국 아래와 같이 개별 position의 score 합으로 나타낼 수가 있다.
ProteinGym

결과
•
ProteinGym DMS assay의 mutation effect 예측 성능을 측정함.
•
Metrics = Spearman’s rank correlation coefficient, AUC, MCC
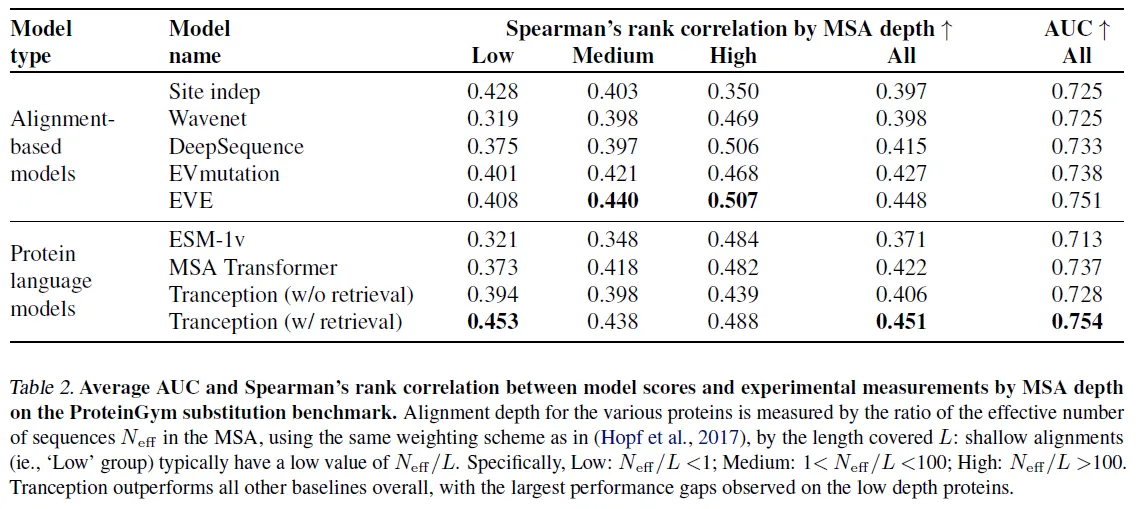
•
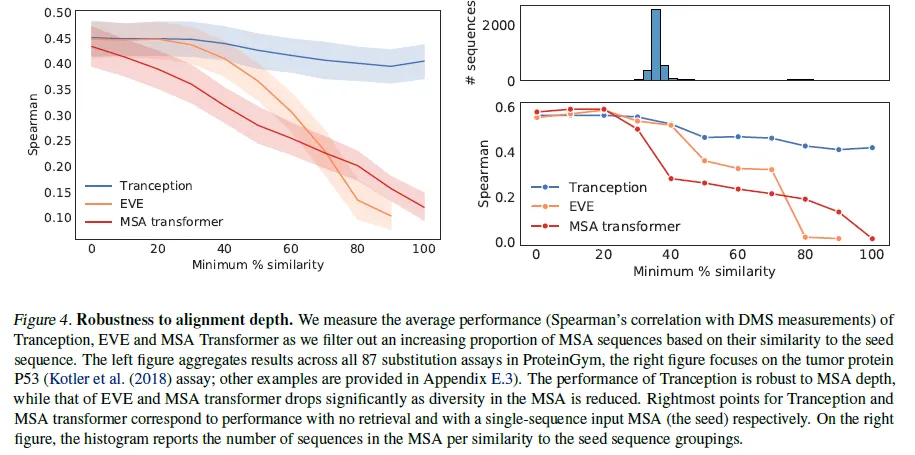
전체적으로 성능 향상이 있으며, MSA depth가 낮을 때 Alignment-based model보다 좋은 성능을 보임에 주목.
•
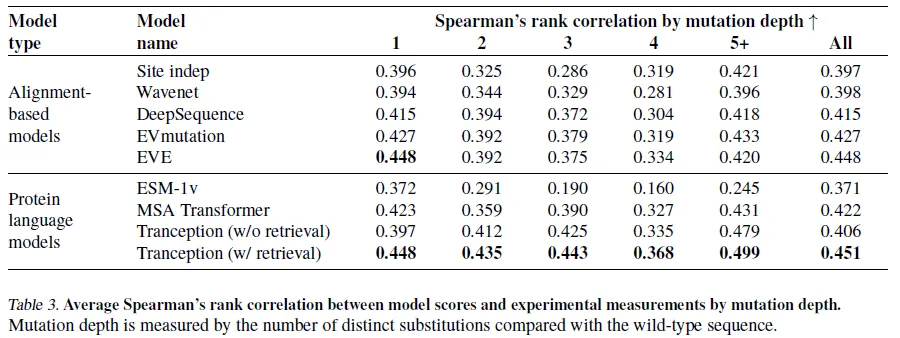
Multiple mutation이 있는 경우에도 성능이 좋음.
•
Sequence % similarity cutoff를 높여가며 MSA depth를 낮추면서 성능이 어떻게 변하는지 측정. MSA 기반의 방법과 비교하면 성능이 조금밖에 안 떨어진다.
결론
•
Tranception은 alignment-free 서열로 학습된 ESM-1v 모델과, MSA를 이용하여 학습된 MSA transformer의 hybrid라고 볼 수 있다.
◦
Alignment-free → Alignable한 단백질에만 국한되지 않는 학습이 가능함
◦
MSA를 학습에 이용하지 않음 → MSA를 만드는 파라미터에 덜 sensitive해짐